Histone Variant H3.3 Mutations in Defining the Chromatin Function in Mammals


Abstract
:1. Introduction
2. Histone H3 Variants
3. Histone Variant H3.3
4. Histone Residue Mutagenesis in Model Organisms: From Yeasts to Drosophila
4.1. Yeast
4.2. Other Fungi
4.3. Fly
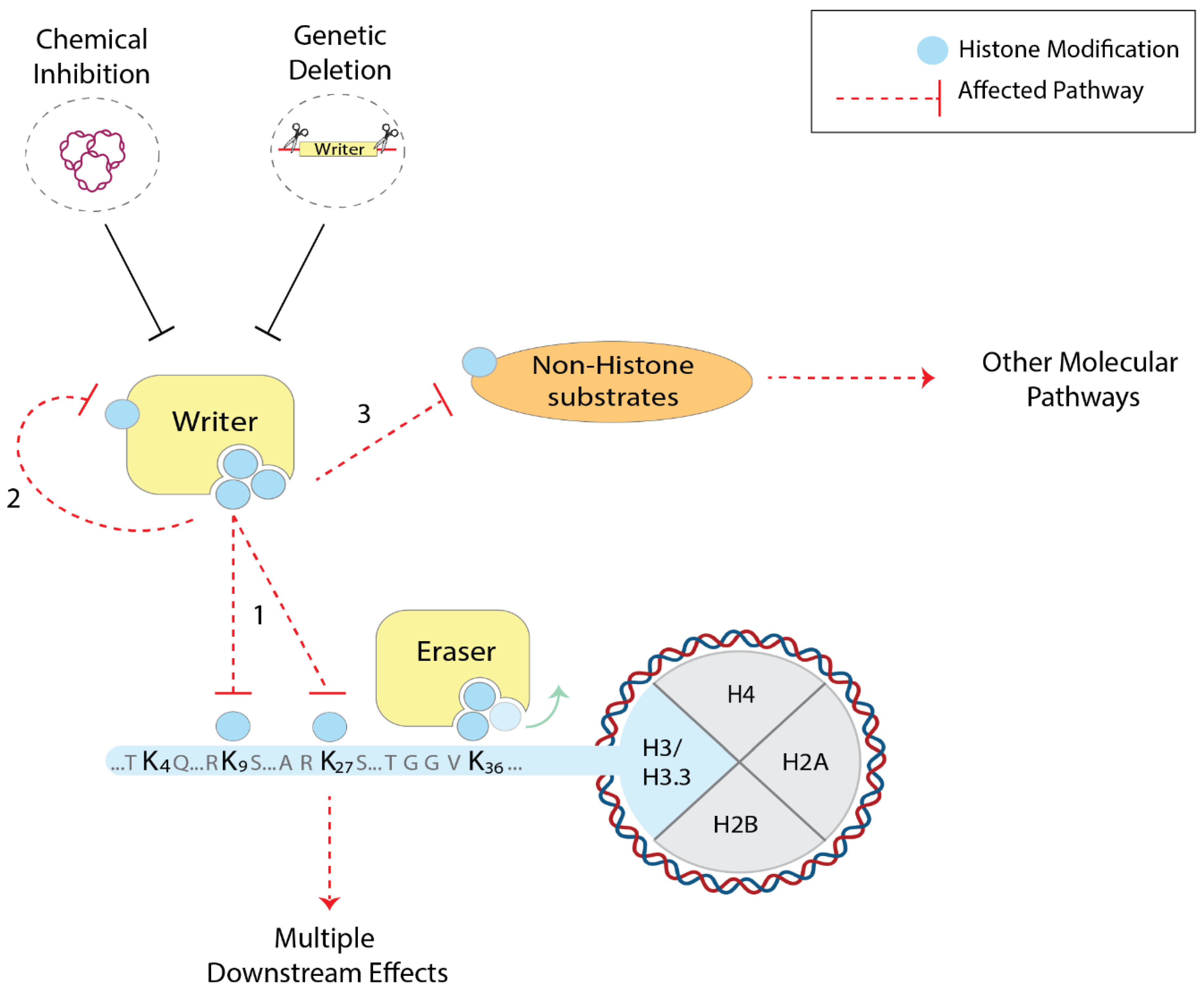
5. Histone PTM Studies through the Perturbation of Chromatin Factors
6. H3.3 Mutations in Cancer
7. Histone H3.3 Mutagenesis: A Way to Study Histone Residues and PTMs in Mammals
8. H3.3 Mutagenesis beyond Cancer
8.1. K9M and K36M
8.2. K4A/R and K36A
8.3. K27R
8.4. S31A/E
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kornberg, R.D.; Thomas, J.O. Chromatin structure: Oligomers of the histones. Science 1974, 184, 865–868. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Bonn, S.; Zinzen, R.P.; Girardot, C.; Gustafson, E.H.; Perez-Gonzalez, A.; Delhomme, N.; Ghavi-Helm, Y.; Wilczyåski, B.; Riddell, A.; Furlong, E.E.M. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musselman, C.A.; Lalonde, M.E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.; Sun, X.; Shi, W.; Yanru, A.; Leung, S.T.C.; Ding, D.; Cheema, M.S.; MacPherson, N.; Nelson, C.J.; Ausio, J.; et al. A novel histone H4 variant H4G regulates rDNA transcription in breast cancer. Nucleic Acids Res. 2019, 47, 8399–8409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef] [PubMed]
- Kamakaka, R.T.; Biggins, S. Histone variants: Deviants? Genes Dev. 2005, 19, 295–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maze, I.; Noh, K.-M.; Soshnev, A.A.; Allis, C.D. Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Marzluff, W.; Gongidi, P.; Woods, K.; Jin, J.; Maltais, L. The Human and Mouse Replication-Dependent Histone Genes. Genomics 2002, 80, 487–498. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Bassett, E.; Chakravarti, A.; Parthun, M.R. Replication-dependent histone isoforms: A new source of complexity in chromatin structure and function. Nucleic Acids Res. 2018, 46, 8665–8678. [Google Scholar] [CrossRef]
- Dominski, Z.; Marzluff, W.F. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene 2007, 396, 373–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, R.; Jenke, A.; Zilbauer, M.; Wirth, S.; Postberg, J. H3.5 is a novel hominid-specific histone H3 variant that is specifically expressed in the seminiferous tubules of human testes. Chromosoma 2011, 120, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Müller, S.; Almouzni, G. Histone H3 Variants and Their Chaperones During Development and Disease: Contributing to Epigenetic Control. Annu. Rev. Cell Dev. Biol. 2014, 30, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, S.M.; Mildner, S.N.; Bönisch, C.; Israel, L.; Maiser, A.; Matheisl, S.; Straub, T.; Merkl, R.; Leonhardt, H.; Kremmer, E.; et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell Biol. 2010, 190, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Howman, E.V.; Fowler, K.J.; Newson, A.J.; Redward, S.; MacDonald, A.C.; Kalitsis, P.; Choo, K.H. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc. Natl. Acad. Sci. USA 2000, 97, 1148–1153. [Google Scholar] [CrossRef] [Green Version]
- Dunleavy, E.; Pidoux, A.; Allshire, R. Centromeric chromatin makes its mark. Trends Biochem. Sci. 2005, 30, 172–175. [Google Scholar] [CrossRef]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- Maze, I.; Wenderski, W.; Noh, K.-M.; Bagot, R.C.; Tzavaras, N.; Purushothaman, I.; Elsässer, S.J.; Guo, Y.; Ionete, C.; Hurd, Y.L.; et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 2015, 87, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Maehara, K.; Harada, A.; Sato, Y.; Matsumoto, M.; Nakayama, K.I.; Kimura, H.; Ohkawa, Y. Tissue-specific expression of histone H3 variants diversified after species separation. Epigenetics Chromatin 2015, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.-W.; Shibata, Y.; Starmer, J.; Yee, D.; Magnuson, T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015, 29, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.C.W.; Jacobs, S.A.; Wong, L.H.; Mann, J.R. Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis 2013, 51, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.C.W.; Jacobs, S.A.; Mattiske, D.M.; Soh, Y.M.; Graham, A.N.; Tran, A.; Lim, S.L.; Hudson, D.F.; Kalitsis, P.; O’Bryan, M.K.; et al. Contribution of the Two Genes Encoding Histone Variant H3.3 to Viability and Fertility in Mice. PLoS Genet. 2015, 11, e1004964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couldrey, C.; Carlton, M.B.L.; Nolan, P.M.; Colledge, W.H.; Evans, M.J. A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum. Mol. Genet. 1999, 8, 2489–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.M.; Yuen, B.T.; Barrilleaux, B.L.; Riggs, J.W.; O’Geen, H.; Cotterman, R.F.; Knoepfler, P.S. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin 2013, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Yuen, B.T.K.; Knoepfler, P.S. Histone H3.3 Mutations: A Variant Path to Cancer. Cancer Cell 2013, 24, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Muhire, B.M.; Booker, M.A.; Tolstorukov, M.Y. Non-neutral evolution of H3.3-encoding genes occurs without alterations in protein sequence. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hake, S.B.; Garcia, B.A.; Kauer, M.; Baker, S.P.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc. Natl. Acad. Sci. USA 2005, 102, 6344–6349. [Google Scholar] [CrossRef] [Green Version]
- Elsaesser, S.J.; Goldberg, A.D.; Allis, C.D. New functions for an old variant: No substitute for histone H3.3. Curr. Opin. Genet. Dev. 2010, 20, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.-M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Pchelintsev, N.A.; McBryan, T.; Rai, T.S.; VanTuyn, J.; Ray-Gallet, D.; Almouzni, G.; Adams, P.D. Placing the HIRA Histone Chaperone Complex in the Chromatin Landscape. Cell Rep. 2013, 3, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Xiong, C.; Wen, Z.; Yu, J.; Chen, J.; Liu, C.P.; Zhang, X.; Chen, P.; Xu, R.M.; Li, G. UBN1/2 of HIRA complex is responsible for recognition and deposition of H3.3 at cis-regulatory elements of genes in mouse ES cells. BMC Biol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.-M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsässer, S.J.; Noh, K.-M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Udugama, M.; Chang, F.T.M.; Chan, F.L.; Tang, M.C.; Pickett, H.A.; McGhie, J.D.R.; Mayne, L.; Collas, P.; Mann, J.R.; Wong, L.H. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 2015, 43, 10227–10237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drané, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.; Mollah, S.; Garcia, B.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F.; Jacobsen, S.E. Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res. 2004, 32, 6511–6518. [Google Scholar] [CrossRef] [Green Version]
- Hake, S.B.; Garcia, B.A.; Duncan, E.M.; Kauer, M.; Dellaire, G.; Shabanowitz, J.; Bazett-Jones, D.P.; Allis, C.D.; Hunt, D.F. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 2006, 281, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Shilatifard, A. Histone modification: Cause or cog? Trends Genet. 2011, 27, 389–396. [Google Scholar] [CrossRef]
- Smith, M.M.; Santisteban, M.S. Genetic dissection of histone function. Methods 1998, 15, 269–281. [Google Scholar] [CrossRef]
- Dai, J.; Hyland, E.M.; Yuan, D.S.; Huang, H.; Bader, J.S.; Boeke, J.D. Probing Nucleosome Function: A Highly Versatile Library of Synthetic Histone H3 and H4 Mutants. Cell 2008, 134, 1066–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Maertens, A.M.; Hyland, E.M.; Dai, J.; Norris, A.; Boeke, J.D.; Bader, J.S. HistoneHits: A database for histone mutations and their phenotypes. Genome Res. 2009, 19, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyland, E.M.; Cosgrove, M.S.; Molina, H.; Wang, D.; Pandey, A.; Cottee, R.J.; Boeke, J.D. Insights into the Role of Histone H3 and Histone H4 Core Modifiable Residues in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005, 25, 10060–10070. [Google Scholar] [CrossRef] [Green Version]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J.Y.; et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015, 29, 1362–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, S.; Sanderson, B.W.; Delventhal, K.M.; Bradford, W.D.; Staehling-Hampton, K.; Shilatifard, A. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 2008, 15, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nützmann, H.W.; Fischer, J.; Scherlach, K.; Hertweck, C.; Brakhagea, A.A. Distinct amino acids of histone H3 control secondary metabolism in aspergillus nidulans. Appl. Environ. Microbiol. 2013, 79, 6102–6109. [Google Scholar] [CrossRef] [Green Version]
- Adhvaryu, K.K.; Berge, E.; Tamaru, H.; Freitag, M.; Selker, E.U. Substitutions in the Amino-Terminal Tail of Neurospora Histone H3 Have Varied Effects on DNA Methylation. PLoS Genet. 2011, 7, e1002423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, D.J.; Klusza, S.; Penke, T.J.R.; Meers, M.P.; Curry, K.P.; McDaniel, S.L.; Malek, P.Y.; Cooper, S.W.; Tatomer, D.C.; Lieb, J.D.; et al. Interrogating the function of metazoan histones using engineered gene clusters. Dev. Cell 2015, 32, 373–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günesdogan, U.; Jäckle, H.; Herzig, A. A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO Rep. 2010, 11, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, X.; Xue, Z.; Li, Y.; Ma, Q.; Ren, X.; Zhang, J.; Yang, S.; Yang, L.; Wu, M.; et al. Probing the Function of Metazoan Histones with a Systematic Library of H3 and H4 Mutants. Dev. Cell 2019, 48, 406–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.-M.; Morgan, M.; Gao, X.; Jackson, J.; Rickels, R.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014, 345, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Pengelly, A.R.; Copur, Ö.; Jäckle, H.; Herzig, A.; Müller, J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor polycomb. Science 2013, 339, 698–699. [Google Scholar] [CrossRef] [Green Version]
- Meers, M.P.; Henriques, T.; Lavender, C.A.; McKay, D.J.; Strahl, B.D.; Duronio, R.J.; Adelman, K.; Matera, A.G. Histone gene replacement reveals a posttranscriptional role for H3K36 in maintaining metazoan transcriptome fidelity. Elife 2017, 6, e23249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The Polycomb-Group GeneEzh2 Is Required for Early Mouse Development. Mol. Cell. Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef] [Green Version]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Denchi, E.L.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The Polycomb Group Protein Suz12 Is Required for Embryonic Stem Cell Differentiation. Mol. Cell. Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.G.; Cuomo, A.; Barozzi, I.; Stützer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Collinson, A.; Collier, A.J.; Morgan, N.P.; Sienerth, A.R.; Chandra, T.; Andrews, S.; Rugg-Gunn, P.J. Deletion of the Polycomb-Group Protein EZH2 Leads to Compromised Self-Renewal and Differentiation Defects in Human Embryonic Stem Cells. Cell Rep. 2016, 17, 2700–2714. [Google Scholar] [CrossRef] [PubMed]
- Lavarone, E.; Barbieri, C.M.; Pasini, D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Garcia, B.A.; Adelman, K.; Correspondence, J.W. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation Loss of Mll3/4 from enhancers reduces Pol II density in bodies of adjacent genes. Mol. Cell 2017, 66, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickels, R.; Herz, H.M.; Sze, C.C.; Cao, K.; Morgan, M.A.; Collings, C.K.; Gause, M.; Takahashi, Y.H.; Wang, L.; Rendleman, E.J.; et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 2017, 49, 1647–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, E.G.; Mohammad, H.P.; Leadem, B.R.; Easwaran, H.; Cai, Y.; Van Neste, L.; Baylin, S.B. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012, 40, 4334–4346. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Chen, S.A.A.; Local, A.; Liu, T.; Qiu, Y.; Dorighi, K.M.; Preissl, S.; Rivera, C.M.; Wang, C.; Ye, Z.; et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018, 28, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Ugarenko, M.; Ozark, P.A.; Wang, J.; Marshall, S.A.; Rendleman, E.J.; Liang, K.; Wang, L.; Zou, L.; Smith, E.R.; et al. DOT1L-controlled cell-fate determination and transcription elongation are independent of H3K79 methylation. Proc. Natl. Acad. Sci. USA 2020, 117, 27365–27373. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [Green Version]
- Fierz, B.; Muir, T.W. Chromatin as an expansive canvas for chemical biology. Nat. Chem. Biol. 2012, 8, 417–427. [Google Scholar] [CrossRef]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jang, H.; Kim, H.; Lee, J.-H.; Kim, S.-T.; Cho, E.-J.; Youn, H.-D. Modulation of lysine methylation in myocyte enhancer factor 2 during skeletal muscle cell differentiation. Nucleic Acids Res. 2014, 42, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ow, J.R.; Palanichamy Kala, M.; Rao, V.K.; Choi, M.H.; Bharathy, N.; Taneja, R. G9a inhibits MEF2C activity to control sarcomere assembly. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, J.; Cannavò, E.; Crabtree, G.W.; Sun, Z.; Diamantopoulou, A.; Thakur, P.; Chang, C.Y.; Cai, Y.; Lomvardas, S.; Takata, A.; et al. Recapitulation and Reversal of Schizophrenia-Related Phenotypes in Setd1a-Deficient Mice. Neuron 2019, 104, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nelson, K.M.; Strasser, J.M.; Barsyte-Lovejoy, D.; Szewczyk, M.M.; Organ, S.; Cuellar, M.; Singh, G.; Shrimp, J.H.; Nguyen, N.; et al. Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat. Commun. 2017, 8, 1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Gessi, M.; Gielen, G.H.; Hammes, J.; Dörner, E.; Zur Mühlen, A.; Waha, A.; Pietsch, T. H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: Possible diagnostic and therapeutic implications? J. Neurooncol. 2013, 112, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 2014, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernohan, K.D.; Grynspan, D.; Ramphal, R.; Bareke, E.; Wang, Y.C.; Nizalik, E.; Ragoussis, J.; Jabado, N.; Boycott, K.M.; Majewski, J.; et al. H3.1 K36M mutation in a congenital-onset soft tissue neoplasm. Pediatr. Blood Cancer 2017, 64, e26633. [Google Scholar] [CrossRef] [PubMed]
- Koelsche, C.; Schrimpf, D.; Tharun, L.; Roth, E.; Sturm, D.; Jones, D.T.W.; Renker, E.-K.; Sill, M.; Baude, A.; Sahm, F.; et al. Histone 3.3 hotspot mutations in conventional osteosarcomas: A comprehensive clinical and molecular characterization of six H3F3A mutated cases. Clin. Sarcoma Res. 2017, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, D.N.; Allis, C.D.; Lu, C. Oncogenic mechanisms of histone H3 mutations. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; David James, C.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- Stafford, J.M.; Lee, C.H.; Voigt, P.; Descostes, N.; Saldana-Meyer, R.; Yu, J.R.; Leroy, G.; Oksuz, O.; Chapman, J.R.; Suarez, F.; et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 2018, 4, 5935–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yu, J.R.; Kumar, S.; Jin, Y.; LeRoy, G.; Bhanu, N.; Kaneko, S.; Garcia, B.A.; Hamilton, A.D.; Reinberg, D. Allosteric Activation Dictates PRC2 Activity Independent of Its Recruitment to Chromatin. Mol. Cell 2018, 70, 422–434.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.U.; Rashoff, A.Q.; Krabbenhoft, S.D.; Hoelper, D.; Do, T.J.; Gibson, T.J.; Lundgren, S.M.; Bondra, E.R.; Deshmukh, S.; Harutyunyan, A.S.; et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 2020, 80, 726–735.e7. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Quezada, M.A.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 2019, 76, 965–980.e12. [Google Scholar] [CrossRef]
- Sarthy, J.F.; Meers, M.P.; Janssens, D.H.; Henikoff, J.G.; Feldman, H.; Paddison, P.J.; Lockwood, C.M.; Vitanza, N.A.; Olson, J.M.; Ahmad, K.; et al. Histone deposition pathways determine the chromatin landscapes of h3.1 and h3.3 k27m oncohistones. Elife 2020, 9, 1–18. [Google Scholar] [CrossRef]
- Krug, B.; De Jay, N.; Harutyunyan, A.S.; Deshmukh, S.; Marchione, D.M.; Guilhamon, P.; Bertrand, K.C.; Mikael, L.G.; McConechy, M.K.; Chen, C.C.L.; et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 2019, 35, 782–797.e8. [Google Scholar] [CrossRef]
- Silveira, A.B.; Kasper, L.H.; Fan, Y.; Jin, H.; Wu, G.; Shaw, T.I.; Zhu, X.; Larson, J.D.; Easton, J.; Shao, Y.; et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019, 137, 637–655. [Google Scholar] [CrossRef]
- Fellenberg, J.; Sähr, H.; Mancarella, D.; Plass, C.; Lindroth, A.M.; Westhauser, F.; Lehner, B.; Ewerbeck, V. Knock-down of oncohistone H3F3A-G34W counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells. Cancer Lett. 2019, 448, 61–69. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Chen, K.Y.; Bush, K.; Klein, R.H.; Cervantes, V.; Lewis, N.; Naqvi, A.; Carcaboso, A.M.; Lechpammer, M.; Knoepfler, P.S. Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Commun. Biol. 2020, 3, 363. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Gan, H.; Wang, H.; Zhou, H.; Zhang, Z. Probe the function of histone lysine 36 methylation using histone H3 lysine 36 to methionine mutant transgene in mammalian cells. Cell Cycle 2017, 16, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef] [Green Version]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stützer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Shi, J.; Shi, X.; Li, W.; Wen, H. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J. Mol. Biol. 2018, 430, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef] [PubMed]
- Voon, H.P.J.; Udugama, M.; Lin, W.; Hii, L.; Law, R.H.P.; Steer, D.L.; Das, P.P.; Mann, J.R.; Wong, L.H. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Brumbaugh, J.; Kim, I.S.; Ji, F.; Huebner, A.J.; Di Stefano, B.; Schwarz, B.A.; Charlton, J.; Coffey, A.; Choi, J.; Walsh, R.M.; et al. Inducible histone K-to-M mutations are dynamic tools to probe the physiological role of site-specific histone methylation in vitro and in vivo. Nat. Cell Biol. 2019, 21, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Gehre, M.; Bunina, D.; Sidoli, S.; Lübke, M.J.; Diaz, N.; Trovato, M.; Garcia, B.A.; Zaugg, J.B.; Noh, K.-M. Lysine 4 of histone H3.3 is required for embryonic stem cell differentiation, histone enrichment at regulatory regions and transcription accuracy. Nat. Genet. 2020, 52, 273–282. [Google Scholar] [CrossRef]
- Gehre, M.; Buccitelli, C.; Diaz, N.; Korbel, J.; Noh, K.-M. Efficient strategies to detect genome editing and integrity in CRISPR-Cas9 engineered ESCs. bioRxiv 2019. Available online: https://www.biorxiv.org/content/10.1101/635151v1 (accessed on 1 November 2020).
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef]
- Kraushaar, D.C.; Chen, Z.; Tang, Q.; Cui, K.; Zhang, J.; Zhao, K. The gene repressor complex NuRD interacts with the histone variant H3.3 at promoters of active genes. Genome Res. 2018, 28, 1646–1655. [Google Scholar] [CrossRef] [Green Version]
- Jeong, K.W.; Kim, K.; Situ, A.J.; Ulmer, T.S.; An, W.; Stallcup, M.R. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 2011, 18, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 2008, 9, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, J.; Wang, Y.; Wang, M.; Long, H.; Liang, D.; Huang, L.; Wen, Z.; Li, W.; Li, X.; et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013, 27, 2109–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraushaar, D.C.; Jin, W.; Maunakea, A.; Abraham, B.; Ha, M.; Zhao, K. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 2013, 14, R121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Zhang, Z.; Dong, Q.; Xiong, J.; Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020, 21, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leatham-Jensen, M.; Uyehara, C.M.; Strahl, B.D.; Matera, A.G.; Duronio, R.J.; McKay, D.J. Lysine 27 of replication-independent histone H3.3 is required for Polycomb target gene silencing but not for gene activation. PLoS Genet. 2019, 15, e1007932. [Google Scholar] [CrossRef]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 2012, 13, 424. [Google Scholar] [CrossRef] [Green Version]
- Pradeepa, M.M.; Grimes, G.R.; Kumar, Y.; Olley, G.; Taylor, G.C.A.; Schneider, R.; Bickmore, W.A. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet. 2016, 48, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Martire, S.; Gogate, A.A.; Whitmill, A.; Tafessu, A.; Nguyen, J.; Teng, Y.-C.; Tastemel, M.; Banaszynski, L.A. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet. 2019, 51, 941–946. [Google Scholar] [CrossRef]
- Sitbon, D.; Boyarchuk, E.; Dingli, F.; Loew, D.; Almouzni, G. Histone variant H3.3 residue S31 is essential for Xenopus gastrulation regardless of the deposition pathway. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armache, A.; Yang, S.; Martínez de Paz, A.; Robbins, L.E.; Durmaz, C.; Cheong, J.Q.; Ravishankar, A.; Daman, A.W.; Ahimovic, D.J.; Klevorn, T.; et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 2020, 583, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.-H.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef]
- Fang, H.T.; El Farran, C.A.; Xing, Q.R.; Zhang, L.F.; Li, H.; Lim, B.; Loh, Y.H. Global H3.3 dynamic deposition defines its bimodal role in cell fate transition. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Hyland, E.M.; Molina, H.; Poorey, K.; Jie, C.; Xie, Z.; Dai, J.; Qian, J.; Bekiranov, S.; Auble, D.T.; Pandey, A.; et al. An evolutionarily “young” lysine residue in histone h3 attenuates transcriptional output in saccharomyces cerevisiae. Genes Dev. 2011, 25, 1306–1319. [Google Scholar] [CrossRef] [Green Version]
- Graves, H.K.; Wang, P.; Lagarde, M.; Chen, Z.; Tyler, J.K. Mutations that prevent or mimic persistent post-translational modifications of the histone H3 globular domain cause lethality and growth defects in Drosophila. Epigenetics Chromatin 2016, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hayes, J.J. Acetylation Mimics within Individual Core Histone Tail Domains Indicate Distinct Roles in Regulating the Stability of Higher-Order Chromatin Structure. Mol. Cell. Biol. 2008, 28, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.R.; Feng, H.; Ghirlando, R.; Kato, H.; Gruschus, J.; Bai, Y. Histone H4 K16Q mutation, an acetylation mimic, causes structural disorder of its n-terminal basic patch in the nucleosome. J. Mol. Biol. 2012, 421, 30–37. [Google Scholar] [CrossRef] [Green Version]
- White, R.H.; Keberlein, M.; Jackson, V. A mutational mimic analysis of histone H3 post-translational modifications: Specific sites influence the conformational state of H3/H4, causing either positive or negative supercoiling of DNA. Biochemistry 2012, 51, 8173–8188. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yang, A.; Lee, S.; Lee, H.W.; Park, C.B.; Park, H.S. Expanding the genetic code of Mus musculus. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, S.J.; Ernst, R.J.; Walker, O.S.; Chin, J.W. Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat. Methods 2016, 13, 158–164. [Google Scholar] [CrossRef]
- Nadal, S.; Raj, R.; Mohammed, S.; Davis, B.G. Synthetic post-translational modification of histones. Curr. Opin. Chem. Biol. 2018, 45, 35–47. [Google Scholar] [CrossRef]
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. Pombe. Elife 2017, 6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mutation | Genes | Type of Cancers | Molecular Outcomes/Mechanisms | References |
|---|---|---|---|---|
| K27M | H3F3A; HIST1H3B | Pediatric Glioblastomas | Inhibition of PRC2; reduction of H3K27me | [86,87,88,89,91] |
| K36M | H3F3B; HIST1H3C | Chondroblastomas | Inhibition of NSD1/2, SETD2 | [90,92,93,116,118] |
| G34R/V | H3F3A | Pediatric Glioblastomas | Affect SETD2 activity (possibly due to steric hindrance); decrease of H3K36me3 and increase of H3K27me3; inhibition of demthylases | [87,90,91,94,122,123,124] |
| G34W/L | H3F3A | Giant Cell Tumors of the Bone | ||
| G34W/R | H3F3A | Osteosarcomas |
| Mutation | Model Systems | Phenotypic Effects | Molecular Outcomes/Mechanisms | References |
|---|---|---|---|---|
| K4A/R | mESCs; in vitro derived neurons; MEFs | Impaired in vitro differentiation | Extensive gene expression changes; affect binding of NuRD and SWI/SNF remodeler complexes; H3.3 depletion at TSSs and enhancers; dysregulation of Pol II activity | [127,130] |
| K9M | mESCs; HSPCs; Mus musculus | Impaired in vitro differentiation; impaired hematopoiesis in vivo | Increased chromatin accessibility; global reduction of H3K9me; inhibition of H3K9-specific HMTs (i.e., G9a or Setdb1) | [97,126] |
| K27R | mESCs | N/A * | Extensive reduction of H3K27ac at enhancers and modest reduction at TSSs | [135] |
| S31A/E | mESCs | N/A * | H3.3S31ph boosts p300 activity at enhancers | [141] |
| K36A | mESCs; in vitro derived neurons | Higher cell density of neuronal networks | Gene expression changes in neurons; global reduction of H3K36me2/3; increase and spread of H3K27me3 | [116,126,127] |
| K36M | mESCs; HSPCs; Mus musculus | impaired in vitro differentiation; poor body condition and early lethality; impaired spermatogenesis, intestine cells differentiation and hematopiesis in vivo | Increased chromatin accessibility; global reduction of H3K36me3; increase and spread of H3K27me3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trovato, M.; Patil, V.; Gehre, M.; Noh, K.M. Histone Variant H3.3 Mutations in Defining the Chromatin Function in Mammals. Cells 2020, 9, 2716. https://doi.org/10.3390/cells9122716
Trovato M, Patil V, Gehre M, Noh KM. Histone Variant H3.3 Mutations in Defining the Chromatin Function in Mammals. Cells. 2020; 9(12):2716. https://doi.org/10.3390/cells9122716
Chicago/Turabian StyleTrovato, Matteo, Vibha Patil, Maja Gehre, and Kyung Min Noh. 2020. "Histone Variant H3.3 Mutations in Defining the Chromatin Function in Mammals" Cells 9, no. 12: 2716. https://doi.org/10.3390/cells9122716
APA StyleTrovato, M., Patil, V., Gehre, M., & Noh, K. M. (2020). Histone Variant H3.3 Mutations in Defining the Chromatin Function in Mammals. Cells, 9(12), 2716. https://doi.org/10.3390/cells9122716