Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Assays in Microtiter Wells
2.4. Microscopy
2.5. Oxygen Consumption
2.6. Overexpression of the Mutant Gene
3. Results
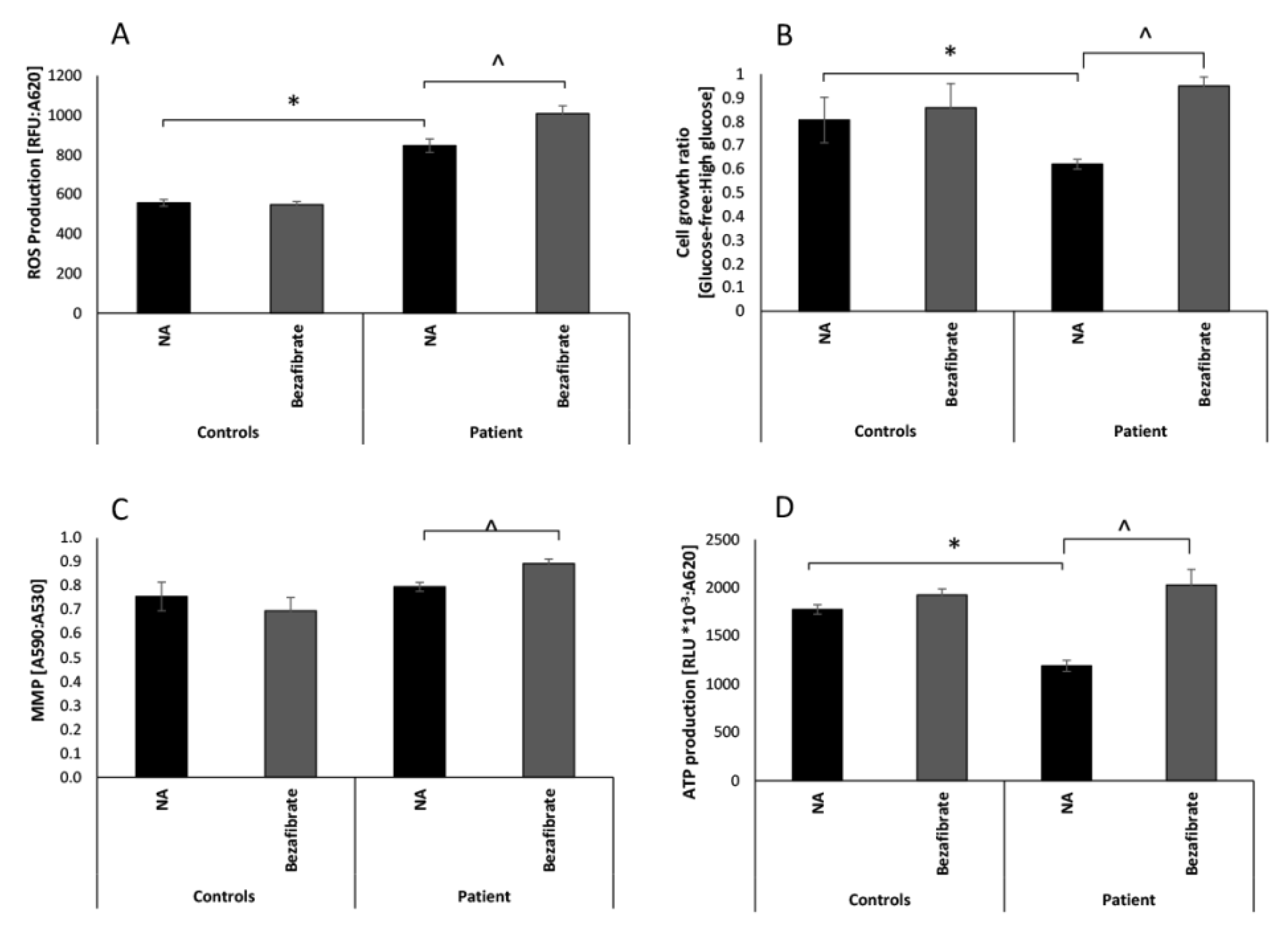
3.1. Bezafibrate Normalizes Growth, ATP Production, and Oxygen Consumption in Patient’s Fibroblasts
3.2. Bezafibrate Improves Mitochondrial Morphology in Patients’ Fibroblasts
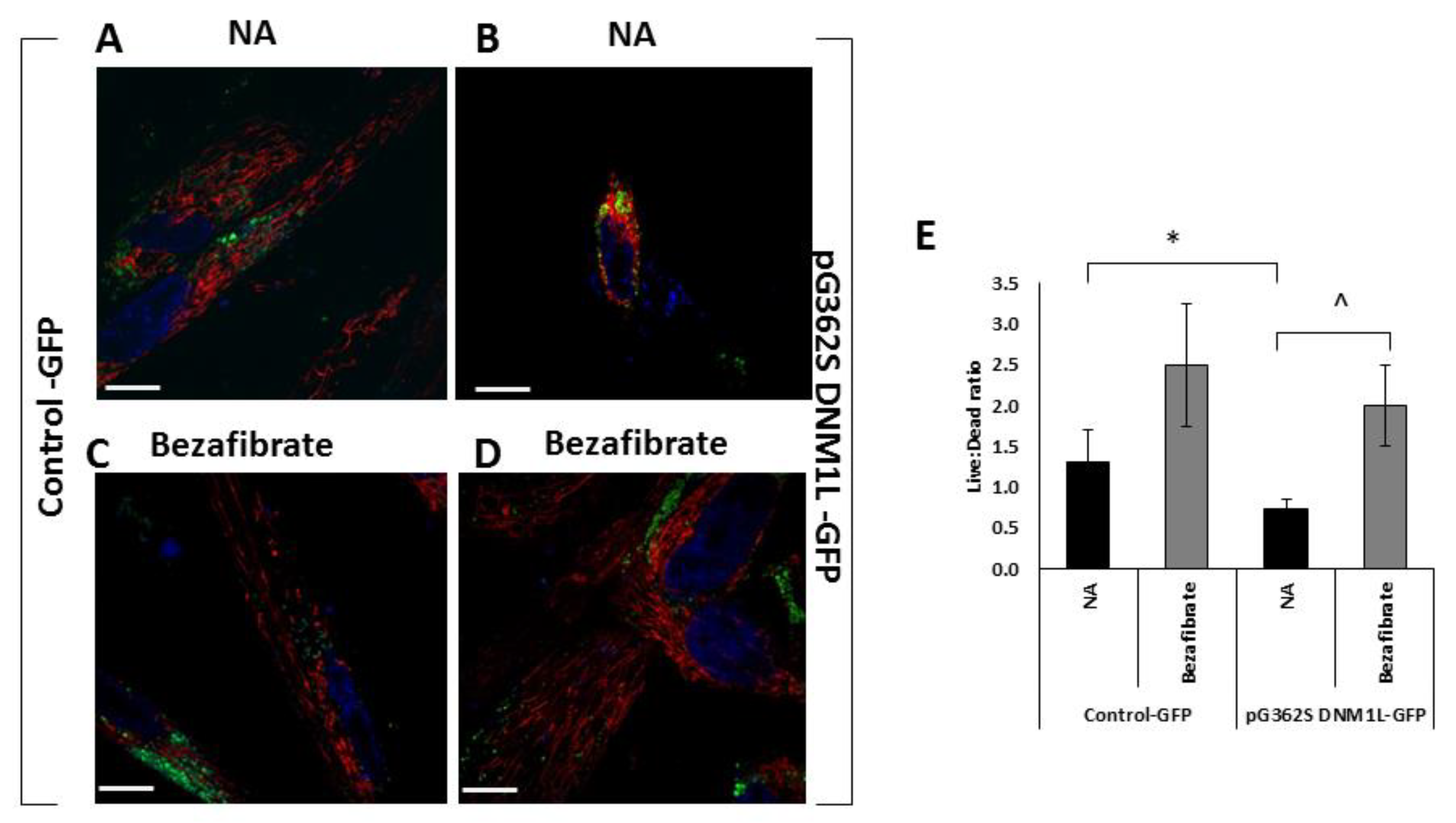
3.3. Bezafibrate Improves Mitochondrial Morphology, Content, and Viability in Fibroblasts Overexpressing the p.G362S DNM1L Mutation
4. Discussion, Limitations, and Conclusions
4.1. Discussion
4.2. Limitations
4.3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvanet, C.; Duvezin-Caubet, S.; di Rago, J.P.; Rojo, M. Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin. Cell Dev. Biol. 2010, 21, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Shirihai, O.; Song, M.; Dorn, G.W., II. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., II. Evolving concepts of mitochondrial dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Chao, Y.H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.J.; Wangler, M.F. AMissense variants in the middle domain of DNM1L in cases of infantile encephalopaty alter peroxisomes and mitochondria when assayed in Drosphila. Hum. Mol. Genet. 2016, 25, 1846–1856. [Google Scholar] [CrossRef] [Green Version]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef]
- Ryan, C.S.; Fine, A.L.; Cohen, A.L.; Schiltz, B.M.; Renaud, D.L.; Wirrell, E.C.; Patterson, M.C.; Boczek, N.J.; Liu, R.; Babovic-Vuksanovic, D.; et al. De novo DNM1L variant in a teenager with progressive paroxysmal dystonia and lethal super-refractory myoclonic status epilepticus. J. Child Neurol. 2018, 33, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum. Mut. 2016, 37, 898–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, G.; Malam, Z.; Paton, T.; Marshall, C.R.; Hyatt, E.; Ivakine, Z.; Scherer, S.W.; Lee, K.S.; Hawkins, C.; Cohn, R.D.; et al. Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J. Pediatr. 2016, 171, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrner, J.A.; Liu, R.; Perry, S.M.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Zaha, K.; Matsumoto, H.; Itoh, M.; Saitsu, H.; Kato, K.; Kato, M.; Ogata, S.; Murayama, K.; Kishita, Y.; Mizuno, Y.; et al. DNM1L-related encephalopathy in infancy with Leigh syndrome-like phenotype and suppression-burst. Clin. Genet. 2016, 90, 472–474. [Google Scholar] [CrossRef]
- Hogarth, K.A.; Costford, S.R.; Yoon, G.; Sondheimer, N.; Maynes, J.T. DNM1L variant alters baseline mitochondrial function and response to stress in a patient with severe neurological dysfunction. Biochemical genetics. Biochem. Genet. 2018, 56, 56–77. [Google Scholar] [CrossRef]
- Ladds, E.; Whitney, A.; Dombi, E.; Hofer, M.; Anand, G.; Harrison, V.; Fratter, C.; Carver, J.; Barbosa, I.A.; Simpson, M.; et al. De novo DNM1L mutation associated with mitochondrial epilepsy syndrome with fever sensitivity. Neurol. Genet. 2018, 4, e258. [Google Scholar] [CrossRef] [Green Version]
- Musto, E.; Gambardella, M.L.; Contaldo, I.; Quintiliani, M.; Perulli, M.; Olivieri, G.; Bompard, S.; Pulitanò, S.; Battini, R.; Bertini, E.; et al. Epileptic Encephalopathy with Recurrent Focal Status Epilepticus and Epilepsia Partialis Continua in Patient with De Novo DNM1L Mutation: Electroclinical Features. Neuropediatrics 2018, 49, S1–S12. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Zahir, F.R.; Zivkovic, I.; Moksa, M.; Selby, K.; Sinha, S.; Nislow, C.; Stockler-Ipsiroglu, S.G.; Sheffer, R.; Saada-Reisch, A.; et al. De novo pathogenic DNM1L variant in a patient diagnosed with atypical hereditary sensory and autonomic neuropathy. Mol. Genet. Genom. Med. 2019, 7, e00961. [Google Scholar] [CrossRef] [Green Version]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999, 1, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van Der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Lightowlers, R.H.; Taylor, R.H.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [Green Version]
- Djouadi, F.; Bastin, J. Mitochondrial genetic disorders: Cell signaling and pharmacological therapies. Cells 2019, 8, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefont, J.P.; Bastin, J.; Behin, A.; Djouadi, F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N. Engl. J. Med. 2009, 360, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for active small molecules in mitochondrial Complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS ONE 2011, 6, e26883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soiferman, D.; Ayalon, O.; Weissman, S.; Saada, A. The effect of small molecules on nuclear-encoded translation diseases. Biochimie 2014, 100, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burté, F.; Saada, A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef]
- Robinson, B.H. Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol. 1996, 264, 454–464. [Google Scholar]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2017, 19, 77–92. [Google Scholar] [CrossRef]
- Saada, A. The use of individual patient’s fibroblasts in the search for personalized treatment of nuclear encoded OXPHOS diseases. Mol. Genet. Metab. 2011, 104, 39–47. [Google Scholar] [CrossRef]
- Saada, A. Mitochondria: Mitochondrial OXPHOS (dys) function ex vivo-The use of primary fibroblasts. Int. J. Biochem. Cell Biol. 2014, 48, 60–65. [Google Scholar] [CrossRef]
- Douiev, L.; Soiferman, D.; Alban, C.; Saada, A. The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. J. Clin. Med. 2016, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Waskowicz, L.R.; Zhou, J.; Landau, D.J.; Brooks, E.D.; Lim, A.; Yavarow, Z.A.; Kudo, T.; Zhang, H.; Wu, Y.; Grant, S.; et al. Bezafibrate induces autophagy and improves hepatic lipid metabolism in glycogen storage disease type Ia. Hum. Mol. Genet. 2019, 28, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douiev, L.; Sheffer, R.; Horvath, G.; Saada, A. Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells. Cells 2020, 9, 301. https://doi.org/10.3390/cells9020301
Douiev L, Sheffer R, Horvath G, Saada A. Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells. Cells. 2020; 9(2):301. https://doi.org/10.3390/cells9020301
Chicago/Turabian StyleDouiev, Liza, Ruth Sheffer, Gabriella Horvath, and Ann Saada. 2020. "Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells" Cells 9, no. 2: 301. https://doi.org/10.3390/cells9020301
APA StyleDouiev, L., Sheffer, R., Horvath, G., & Saada, A. (2020). Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells. Cells, 9(2), 301. https://doi.org/10.3390/cells9020301