Loading of Primary Human T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation after Polyclonal Stimulation

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanoparticle Synthesis
2.3. Nanoparticle Characterization
2.4. Cell Isolation
2.5. Cell Toxicity
2.6. Cellular Uptake
2.7. Magnetizability
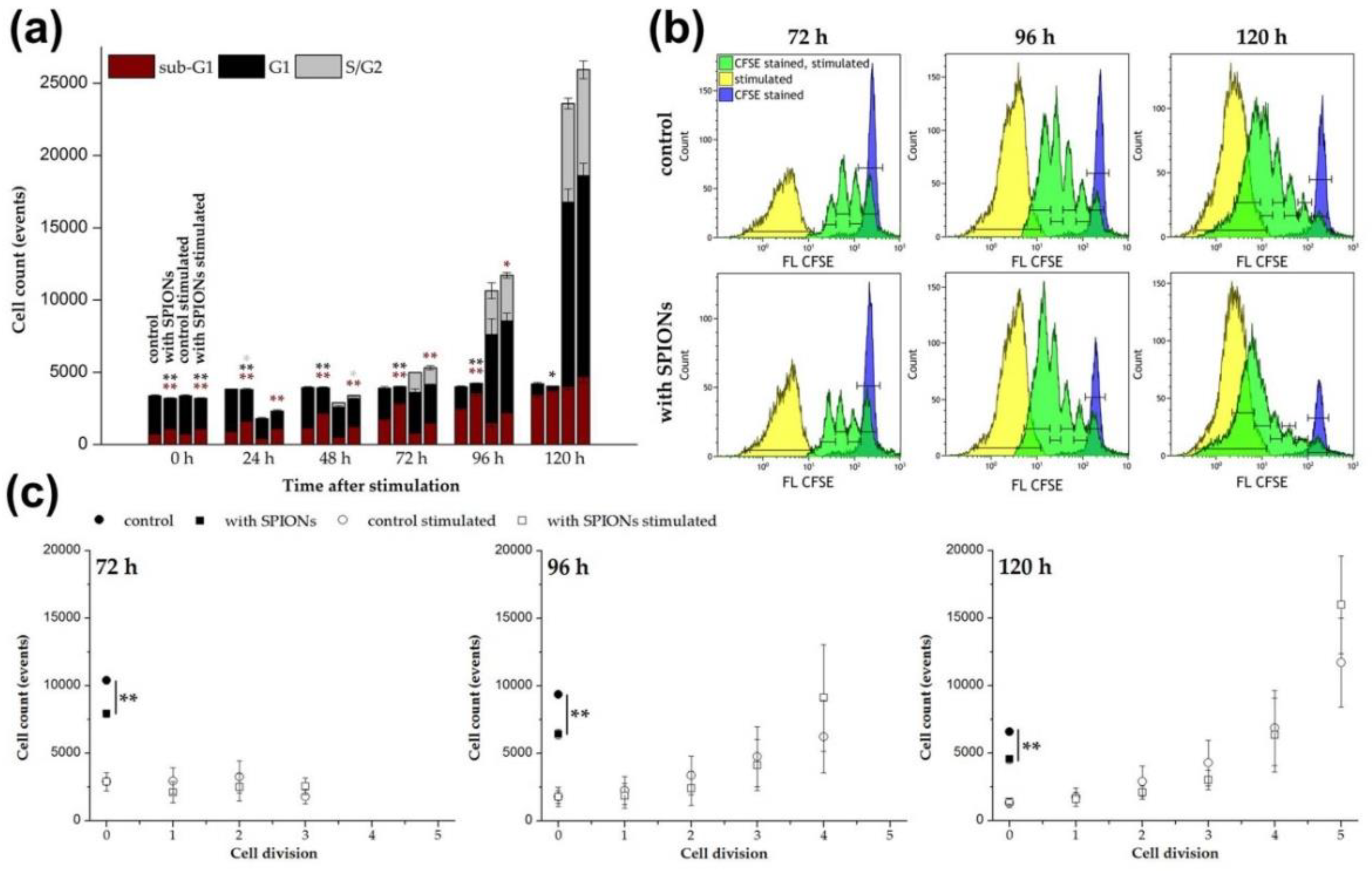
2.8. T Cell Proliferation and Cell Cycle
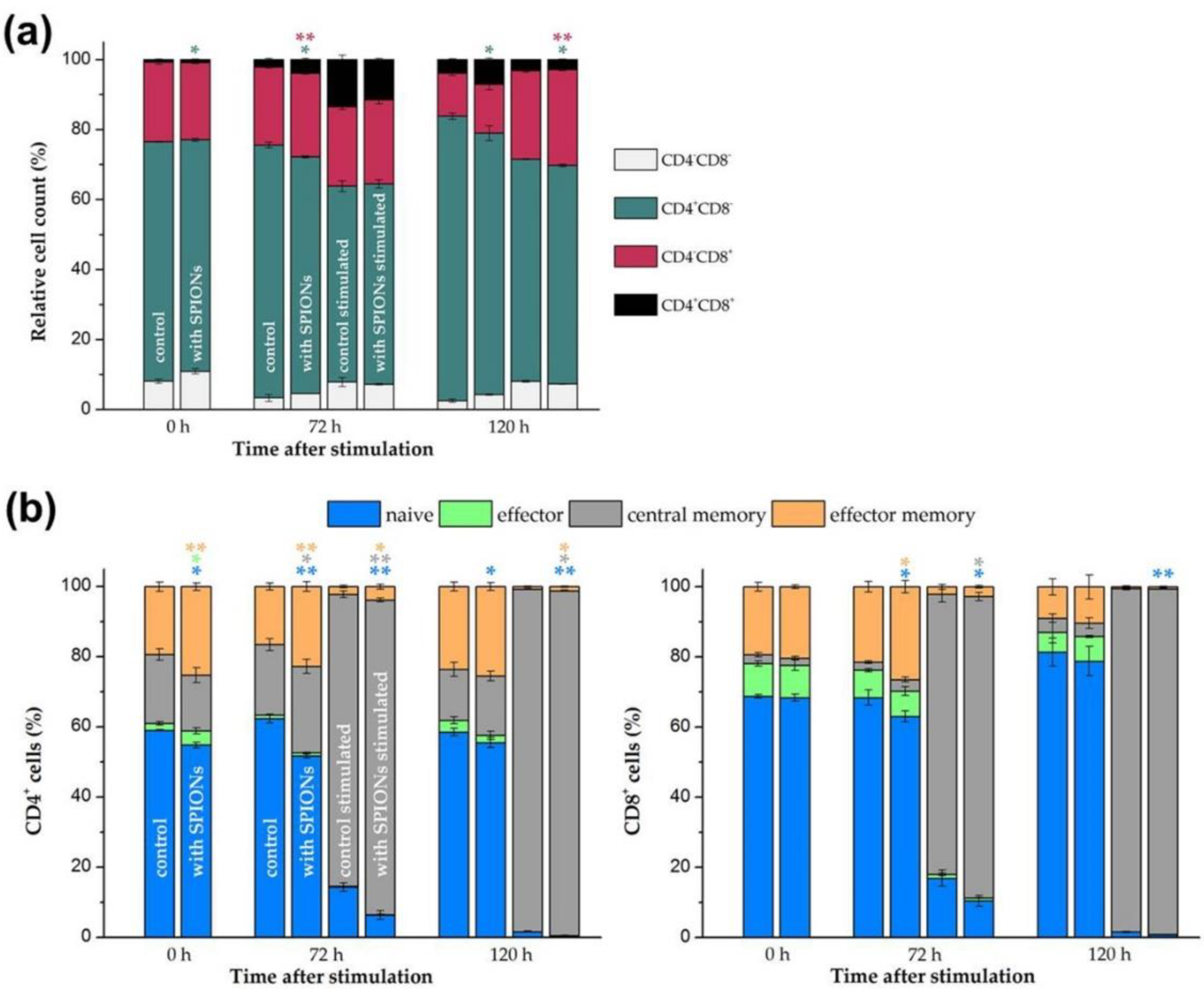
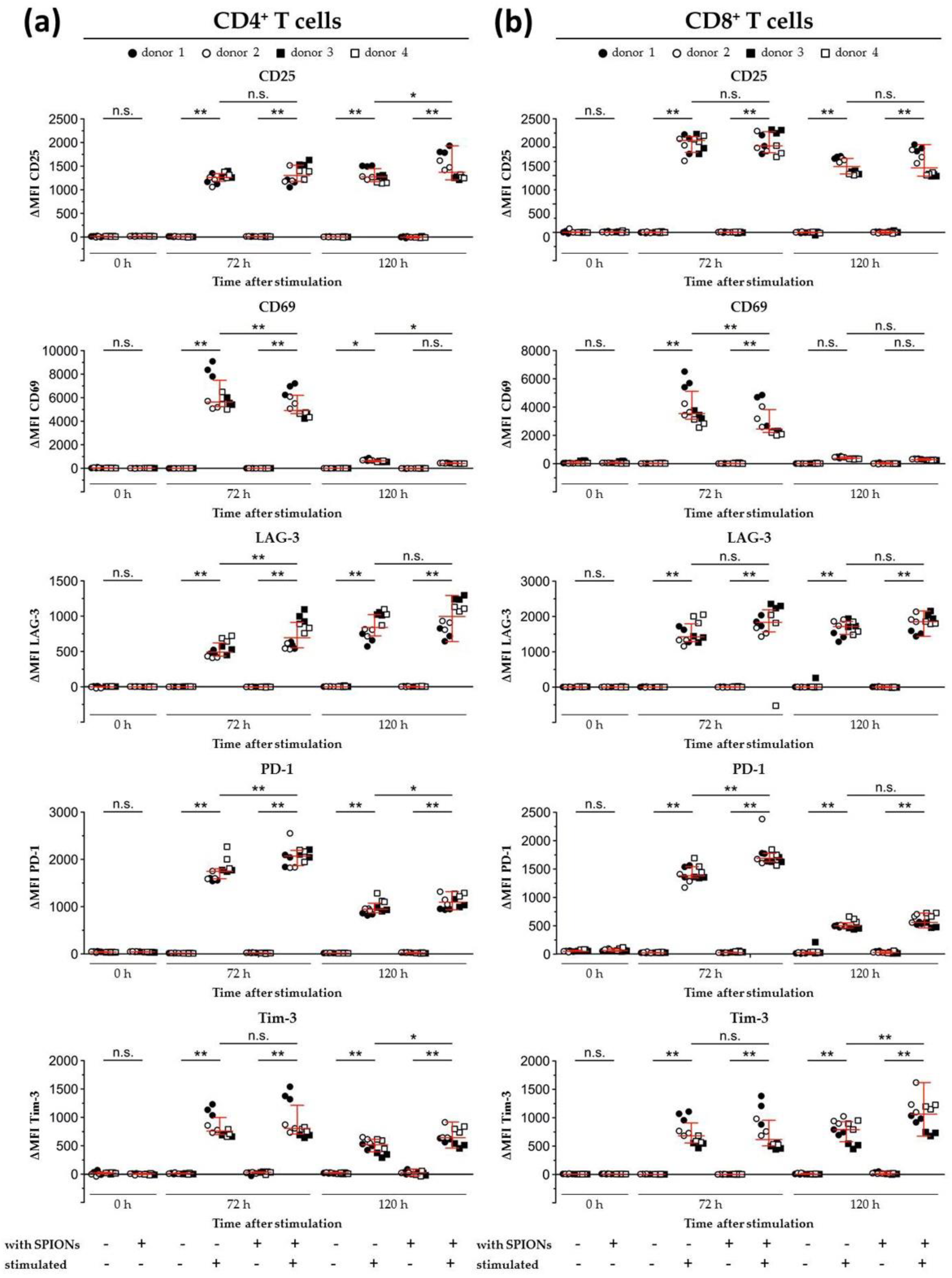
2.9. T Cell Subsets and Activation Markers
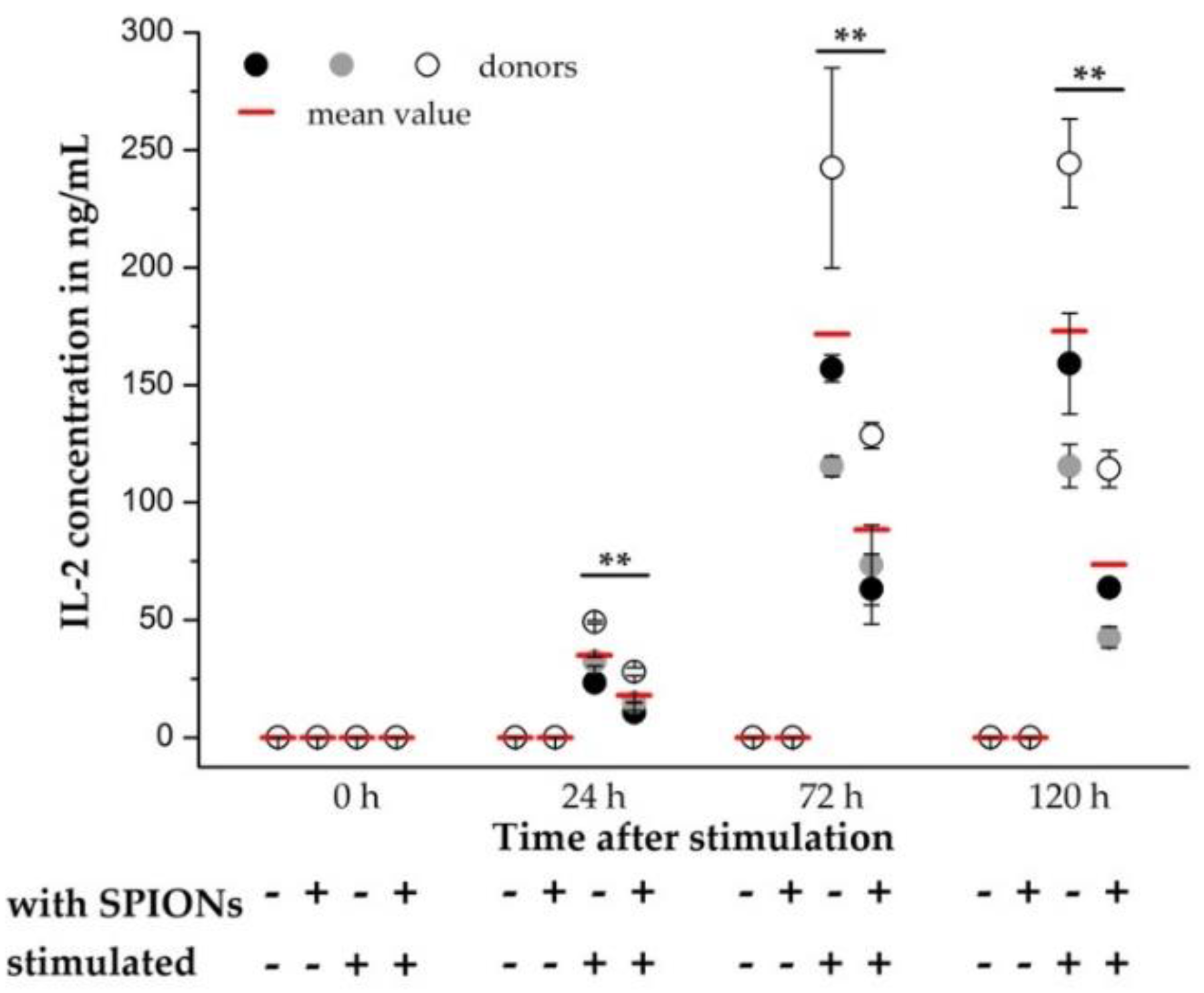
2.10. IL-2 Release
2.11. Statistical Analysis
3. Results and Discussion
3.1. Nanoparticle Characterization
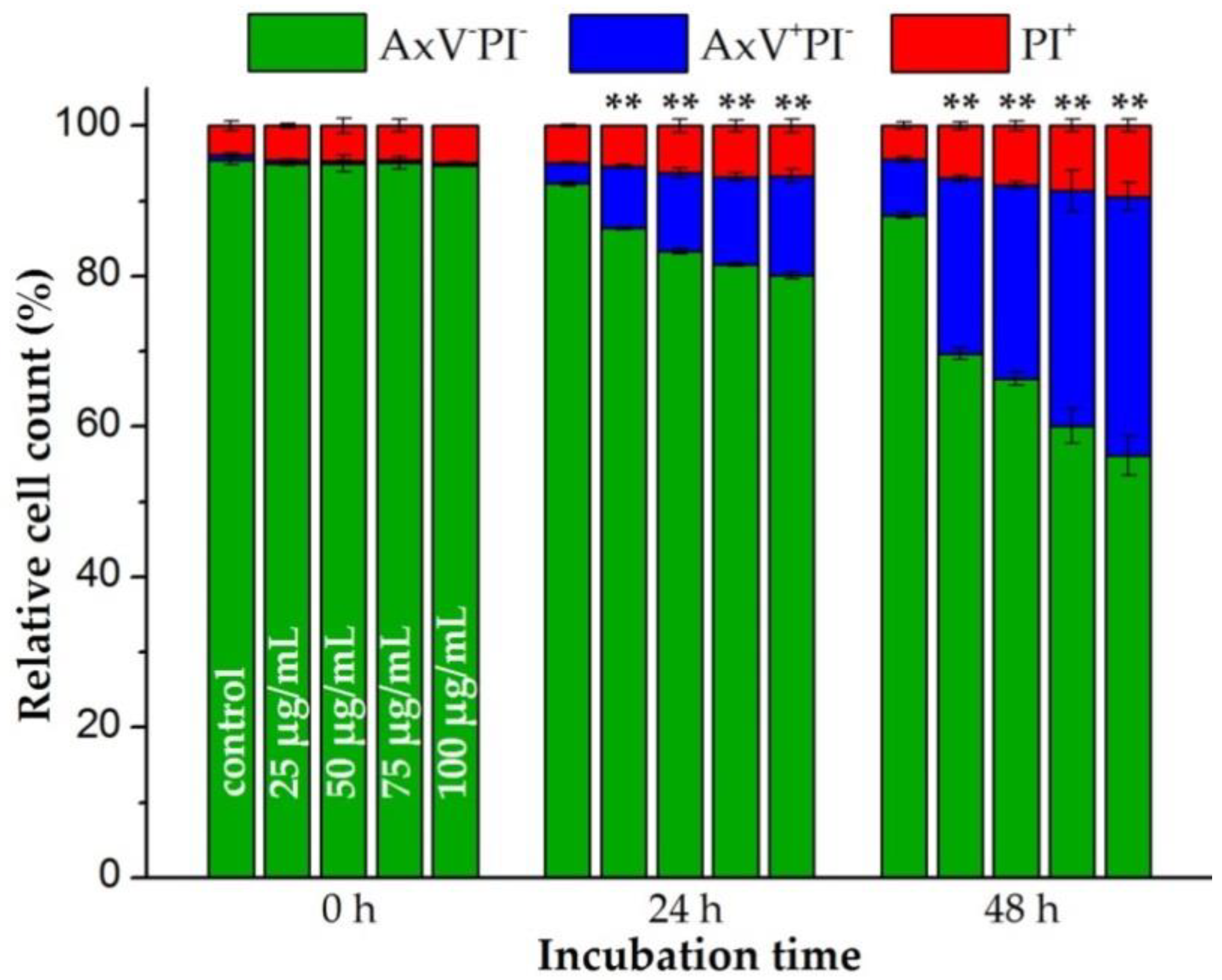
3.2. SPIONCitrate Loading Does Not Have Major Effects on Primary Human T Cell Viability
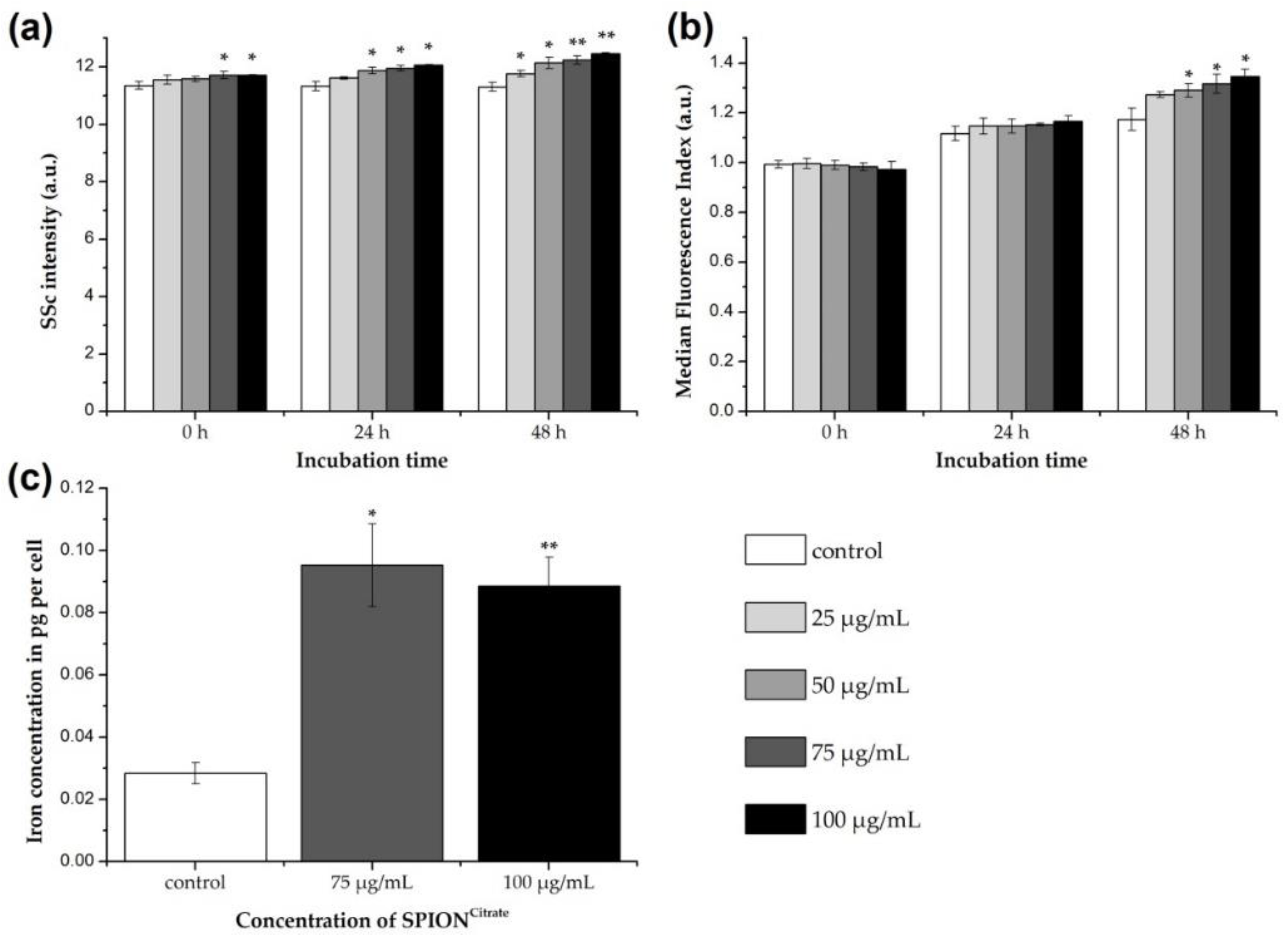
3.3. Cellular Content of SPIONCitrate
3.4. Magnetically Controllable T Cells
3.5. Effects of SPIONCitrate on T Cell Activation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. Ca Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Jass, J.R. Lymphocytic infiltration and survival in rectal cancer. J. Clin. Pathol. 1986, 39, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.-A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef]
- Carreau, N.A.; Pavlick, A.C. Nivolumab and ipilimumab: Immunotherapy for treatment of malignant melanoma. Future Oncol. 2019, 15, 349–358. [Google Scholar] [CrossRef]
- Saruwatari, K.; Sato, R.; Nakane, S.; Sakata, S.; Takamatsu, K.; Jodai, T.; Mito, R.; Horio, Y.; Saeki, S.; Tomita, Y.; et al. The Risks and Benefits of Immune Checkpoint Blockade in Anti-AChR Antibody-Seropositive Non-Small Cell Lung Cancer Patients. Cancers 2019, 11, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009, 69, 4941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Benjamin, J.; Kischel, R.; Stienen, S.; Zugmaier, G. Harnessing T cells to fight cancer with BiTE® antibody constructs – past developments and future directions. Immunol. Rev. 2016, 270, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, M.; D’Argouges, S.; Lorenczewski, G.; Brischwein, K.; Kischel, R.; Lutterbuese, R.; Mangold, S.; Rau, D.; Volkland, J.; Pflanz, S.; et al. Antitumor activity of an EpCAM/CD3-bispecific BiTE antibody during long-term treatment of mice in the absence of T-cell anergy and sustained cytokine release. J. Immunother. 2009, 32, 452–464. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Khadka, R.H.; Sakemura, R.; Kenderian, S.S.; Johnson, A.J. Management of cytokine release syndrome: An update on emerging antigen-specific T cell engaging immunotherapies. Immunotherapy 2019, 11, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Vanecek, V.; Zablotskii, V.; Forostyak, S.; Ruzicka, J.; Herynek, V.; Babic, M.; Jendelova, P.; Kubinova, S.; Dejneka, A.; Sykova, E. Highly efficient magnetic targeting of mesenchymal stem cells in spinal cord injury. Int. J. Nanomed. 2012, 7, 3719–3730. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Huang, N.; Ma, B.; Maitz, M.F.; Wang, J.; Li, J.; Li, Q.; Zhao, Y.; Xiong, K.; Liu, X. Guidance of Stem Cells to a Target Destination in Vivo by Magnetic Nanoparticles in a Magnetic Field. Acs Appl. Mater. Interfaces 2013, 5, 5976–5985. [Google Scholar] [CrossRef]
- Cores, J.; Caranasos, T.G.; Cheng, K. Magnetically Targeted Stem Cell Delivery for Regenerative Medicine. J. Funct. Biomater. 2015, 6, 526–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goya, G.F.; Marcos-Campos, I.; Fernandez-Pacheco, R.; Saez, B.; Godino, J.; Asin, L.; Lambea, J.; Tabuenca, P.; Mayordomo, J.I.; Larrad, L.; et al. Dendritic cell uptake of iron-based magnetic nanoparticles. Cell Biol. Int. 2008, 32, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ortega, L.; Rojas, J.M.; Portilla, Y.; Perez-Yague, S.; Barber, D.F. Magnetic Nanoparticles Attached to the NK Cell Surface for Tumor Targeting in Adoptive Transfer Therapies Does Not Affect Cellular Effector Functions. Front. Immunol. 2019, 10, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlberger, M.; Janko, C.; Unterweger, H.; Schreiber, E.; Band, J.; Lehmann, C.; Dudziak, D.; Lee, G.; Alexiou, C.; Tietze, R. Functionalization of T lymphocytes for magnetically controlled immune therapy: Selection of suitable superparamagnetic iron oxide nanoparticles. J. Magn. Magn. Mater. 2019, 473, 61–67. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Rojas, J.M.; Marcos, A.; Portilla, Y.; Stein, J.V.; Barber, D.F. T cells loaded with magnetic nanoparticles are retained in peripheral lymph nodes by the application of a magnetic field. J. Nanobiotechnol. 2019, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Mühlberger, M.; Janko, C.; Unterweger, H.; Friedrich, R.P.; Friedrich, B.; Band, J.; Cebulla, N.; Alexiou, C.; Dudziak, D.; Lee, G.; et al. Functionalization of T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles for Magnetically Controlled Immune Therapy. Int. J. Nanomed. 2019, 14, 8421–8432. [Google Scholar] [CrossRef] [Green Version]
- Elbialy, N.S.; Fathy, M.M.; Khalil, W.M. Doxorubicin loaded magnetic gold nanoparticles for in vivo targeted drug delivery. Int. J. Pharm. 2015, 490, 190–199. [Google Scholar] [CrossRef]
- Lugert, S.; Unterweger, H.; Muhlberger, M.; Janko, C.; Draack, S.; Ludwig, F.; Eberbeck, D.; Alexiou, C.; Friedrich, R.P. Cellular effects of paclitaxel-loaded iron oxide nanoparticles on breast cancer using different 2D and 3D cell culture models. Int. J. Nanomed. 2019, 14, 161–180. [Google Scholar] [CrossRef] [Green Version]
- Bossuyt, X.; Marti, G.E.; Fleisher, T.A. Comparative analysis of whole blood lysis methods for flow cytometry. Cytometry 1997, 30, 124–133. [Google Scholar] [CrossRef]
- Munoz, L.E.; Maueroder, C.; Chaurio, R.; Berens, C.; Herrmann, M.; Janko, C. Colourful death: Six-parameter classification of cell death by flow cytometry--dead cells tell tales. Autoimmunity 2013, 46, 336–341. [Google Scholar] [CrossRef]
- Friedrich, R.P.; Janko, C.; Poettler, M.; Tripal, P.; Zaloga, J.; Cicha, I.; Durr, S.; Nowak, J.; Odenbach, S.; Slabu, I.; et al. Flow cytometry for intracellular SPION quantification: Specificity and sensitivity in comparison with spectroscopic methods. Int. J. Nanomed. 2015, 10, 4185–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quah, B.J.; Warren, H.S.; Parish, C.R. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007, 2, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.A.; Yirinec, B.D.; Silverstein, S.C. Phorbol esters and horseradish peroxidase stimulate pinocytosis and redirect the flow of pinocytosed fluid in macrophages. J. Cell Biol. 1985, 100, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Hanani, M. Lucifer yellow — An angel rather than the devil. J. Cell. Mol. Med. 2012, 16, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Marsh, M.; McMahon, H.T. The Structural Era of Endocytosis. Science 1999, 285, 215. [Google Scholar] [CrossRef] [Green Version]
- Waiczies, S.; Niendorf, T.; Lombardi, G. Labeling of cell therapies: How can we get it right? Oncoimmunology 2017, 6, e1345403. [Google Scholar] [CrossRef] [Green Version]
- Renner, K.; Bruss, C.; Färber, S. Monitoring Human T Cell Activation in the Context of Immunotherapeutic Approaches: Determination of Cell Growth and Proliferation using CASY. Available online: https://www.ols-bio.de/media/pdf/T-cell-activation-CASY-AppNote_OLS.pdf (accessed on 30 January 2020).
- Ueda, Y.; Ishiwata, T.; Shinji, S.; Arai, T.; Matsuda, Y.; Aida, J.; Sugimoto, N.; Okazaki, T.; Kikuta, J.; Ishii, M.; et al. In vivo imaging of T cell lymphoma infiltration process at the colon. Sci. Rep. 2018, 8, 3978. [Google Scholar] [CrossRef] [Green Version]
- Hennig, T.L.; Unterweger, H.; Lyer, S.; Alexiou, C.; Cicha, I. Magnetic Accumulation of SPIONs under Arterial Flow Conditions: Effect of Serum and Red Blood Cells. Molecules 2019, 24, 2588. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayroldi, E.; Migliorati, G.; Cannarile, L.; Moraca, R.; Delfino, D.V.; Riccardi, C. CD2 Rescues T Cells From T-Cell Receptor/CD3 Apoptosis: A Role for the Fas/Fas-L System. Blood 1997, 89, 3717. [Google Scholar] [CrossRef] [PubMed]
- Green, L. Development of an anti-CD2/CD3/CD28 bead-based T-cell proliferation assay. Biosci. Horiz. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Lier, R.A.W.V.; Brouwer, M.; Aarden, L.A. Signals involved in T cell activation. T cell proliferation induced through the synergistic action of anti-CD28 and anti-CD2 monoclonal antibodies. Eur. J. Immunol. 1988, 18, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Kalland, M.E.; Oberprieler, N.G.; Vang, T.; Tasken, K.; Torgersen, K.M. T cell-signaling network analysis reveals distinct differences between CD28 and CD2 costimulation responses in various subsets and in the MAPK pathway between resting and activated regulatory T cells. J. Immunol. 2011, 187, 5233–5245. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Cooper, G.M. The eukaryotic cell cycle. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000; p. 689. [Google Scholar]
- Lyons, A.B.; Parish, C.R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods 1994, 171, 131–137. [Google Scholar] [CrossRef]
- Parish, C.R. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol. Cell Biol. 1999, 77, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Quah, B.J.; Parish, C.R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 2010, e2259. [Google Scholar] [CrossRef] [Green Version]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Picker, L.J.; Treer, J.R.; Ferguson-Darnell, B.; Collins, P.A.; Buck, D.; Terstappen, L.W. Control of lymphocyte recirculation in man. I. Differential regulation of the peripheral lymph node homing receptor L-selectin on T cells during the virgin to memory cell transition. J. Immunol. 1993, 150, 1105–1121. [Google Scholar] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Liu, X.; Liu, W.X.; Tan, X.Q.; Xiong, F.; Gu, N.; Hao, W.; Gao, X.; Cao, J.M. Fe2O3 nanoparticles suppress Kv1.3 channels via affecting the redox activity of Kvbeta2 subunit in Jurkat T cells. Nanotechnology 2015, 26, 505103. [Google Scholar] [CrossRef] [PubMed]
- Aversa, G.G.; Hall, B.M. Cell surface markers of T-cell activation. Transplant. Rev. 1991, 5, 9–30. [Google Scholar] [CrossRef]
- Bajnok, A.; Ivanova, M.; Rig ó, J.; Toldi, G. The Distribution of Activation Markers and Selectins on Peripheral T Lymphocytes in Preeclampsia. Mediat. Inflamm. 2017, 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Ramsdell, F.; Alderson, M.R. The activation antigen CD69. Stem Cells 1994, 12, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Létourneau, S.; Krieg, C.; Pantaleo, G.; Boyman, O. IL-2– and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J. Allergy Clin. Immunol. 2009, 123, 758–762. [Google Scholar] [CrossRef]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef]
- Puhr, H.C.; Ilhan-Mutlu, A. New emerging targets in cancer immunotherapy: The role of LAG3. ESMO Open 2019, 4, e000482. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, H.; Kane, L.P. Immune regulation by Tim-3. F1000Res 2018, 7, 316. [Google Scholar] [CrossRef] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallett, G.; Laurence, A.; Amarnath, S. Programmed Cell Death-1 Receptor (PD-1)-Mediated Regulation of Innate Lymphoid Cells. Int. J. Mol. Sci. 2019, 20, 2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ortega, L.; Portilla, Y.; Perez-Yague, S.; Barber, D.F. Magnetic targeting of adoptively transferred tumour-specific nanoparticle-loaded CD8(+) T cells does not improve their tumour infiltration in a mouse model of cancer but promotes the retention of these cells in tumour-draining lymph nodes. J. Nanobiotechnol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorescence | Antibody | Clone | Isotype Control | Clone |
|---|---|---|---|---|
| FITC | anti-human CD3 | UCHT1 | mouse IgG1, κ | MOPC-21 |
| PerCP-Cy5.5 | anti-human CD4 | OKT4 | mouse IgG2b, κ | MPC-11 |
| Pacific Blue | anti-human CD8 | HIT8a | mouse IgG1, κ | MOPC-21 |
| PE | anti-human CD197 (CCR7) | G043H7 | mouse IgG2a, κ | MOPC-173 |
| APC | anti-human CD45RO | UCHL1 | mouse IgG2a, κ | MOPC-173 |
| Fluorescence | Antibody | Clone | Isotype Control | Clone |
|---|---|---|---|---|
| BUV395 | anti-human CD3 | UCHT1 | - | |
| PE | anti-human CD4 | RPA-T4 | - | |
| BUV 737 | anti-human CD8 | SK1 | - | |
| BV 650 | anti-human CD279 (PD-1) | EH12.2H7 | mouse IgG1, κ | MOPC-21 |
| BV 711 | anti-human CD223 (LAG-3) | 11C3C65 | mouse IgG1, κ | MOPC-21 |
| PE/Dazzle594 | anti-human CD366 (Tim-3) | F38-2E2 | mouse IgG1, κ | MOPC-21 |
| PerCP-Cy5.5 | anti-human CD25 | BC96 | mouse IgG1, κ | P3.6.2.8.1 |
| PE-Cy7 | anti-human CD69 | FN50 | mouse IgG1, κ | MOPC-21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mühlberger, M.; Unterweger, H.; Band, J.; Lehmann, C.; Heger, L.; Dudziak, D.; Alexiou, C.; Lee, G.; Janko, C. Loading of Primary Human T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation after Polyclonal Stimulation. Cells 2020, 9, 342. https://doi.org/10.3390/cells9020342
Mühlberger M, Unterweger H, Band J, Lehmann C, Heger L, Dudziak D, Alexiou C, Lee G, Janko C. Loading of Primary Human T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation after Polyclonal Stimulation. Cells. 2020; 9(2):342. https://doi.org/10.3390/cells9020342
Chicago/Turabian StyleMühlberger, Marina, Harald Unterweger, Julia Band, Christian Lehmann, Lukas Heger, Diana Dudziak, Christoph Alexiou, Geoffrey Lee, and Christina Janko. 2020. "Loading of Primary Human T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation after Polyclonal Stimulation" Cells 9, no. 2: 342. https://doi.org/10.3390/cells9020342
APA StyleMühlberger, M., Unterweger, H., Band, J., Lehmann, C., Heger, L., Dudziak, D., Alexiou, C., Lee, G., & Janko, C. (2020). Loading of Primary Human T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation after Polyclonal Stimulation. Cells, 9(2), 342. https://doi.org/10.3390/cells9020342