SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Osteosarcoma and Ewing Sarcoma
2. Current Main Treatments and Perspectives
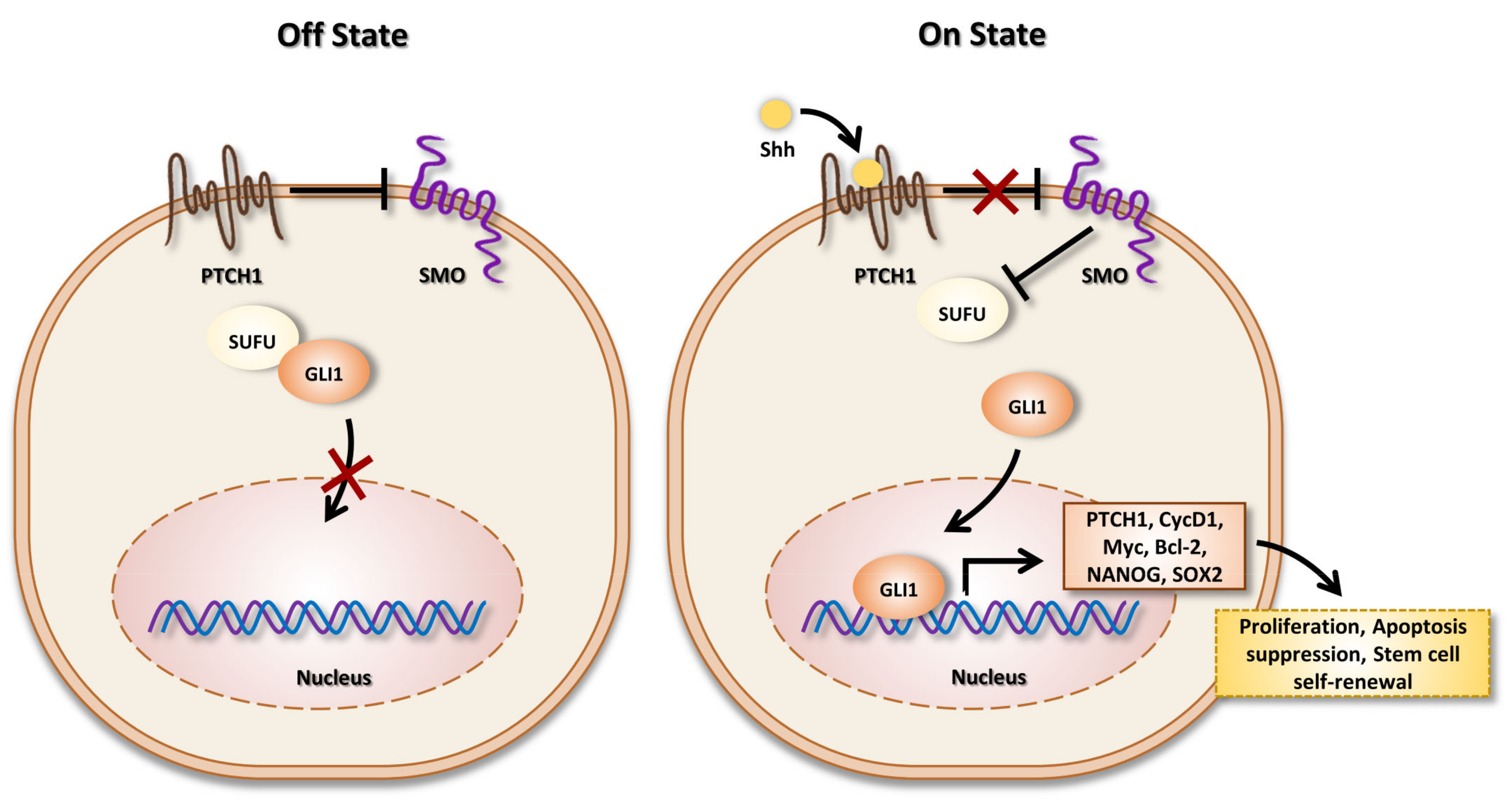
3. SHH Signaling Pathway
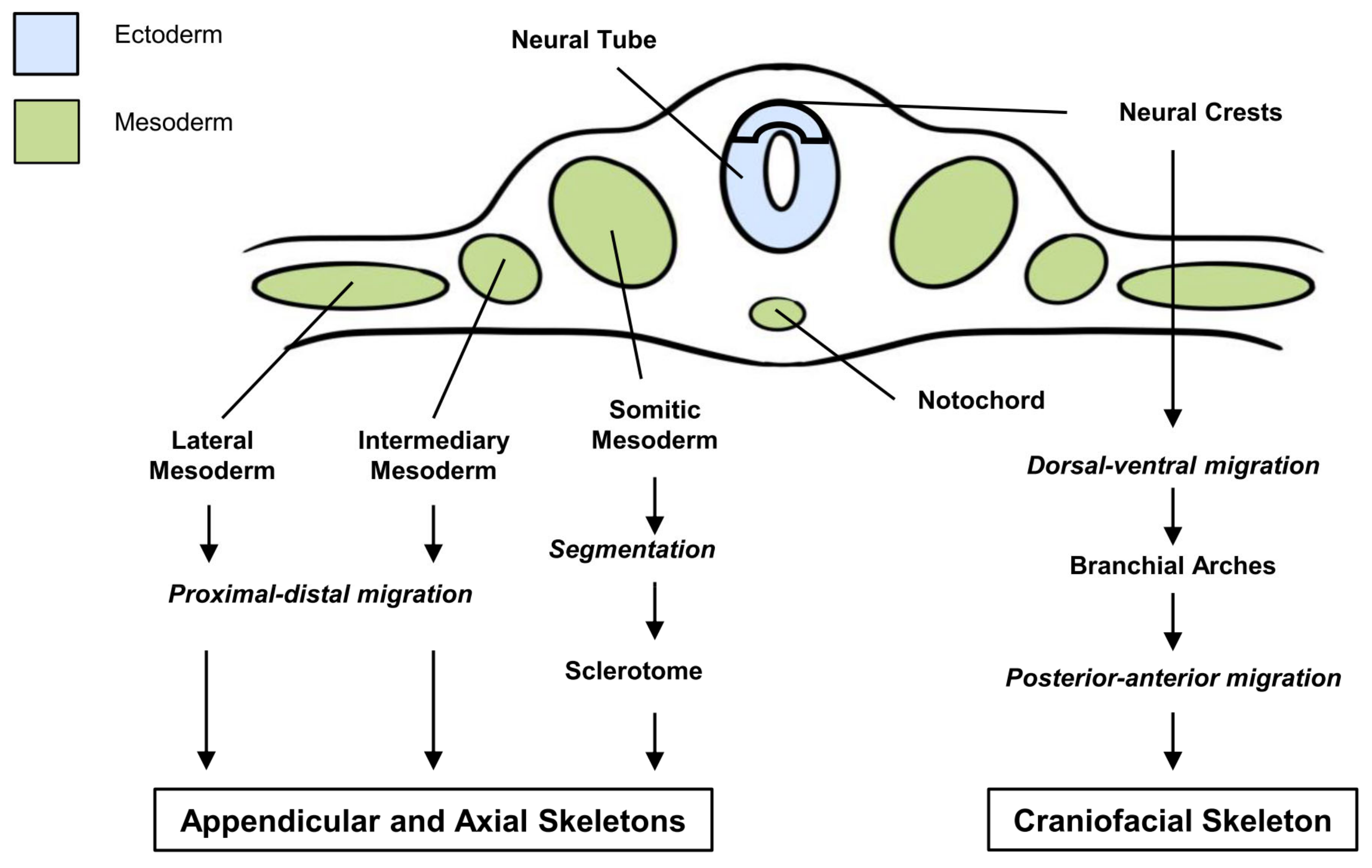
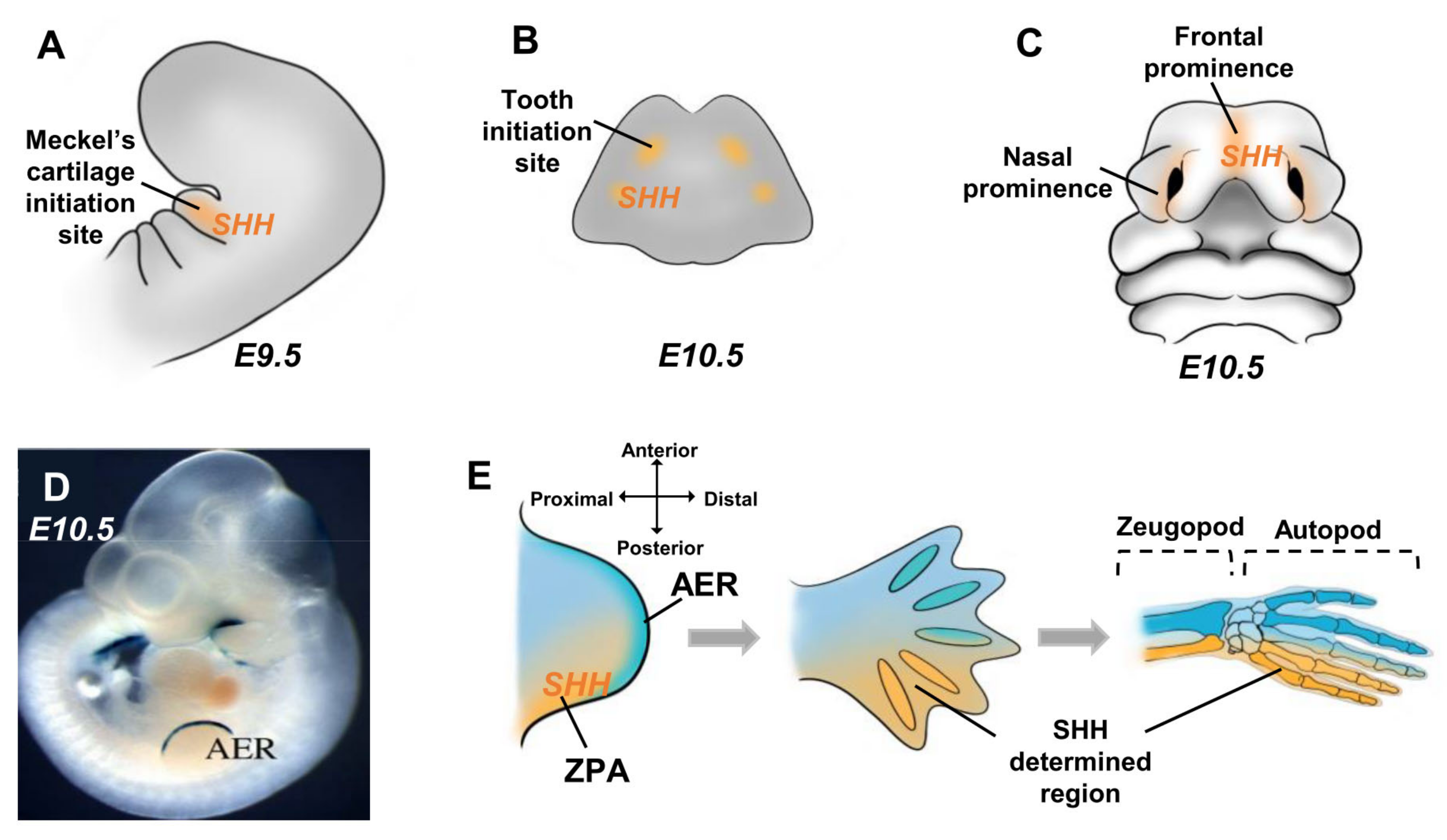
4. SHH and Skeletal Development
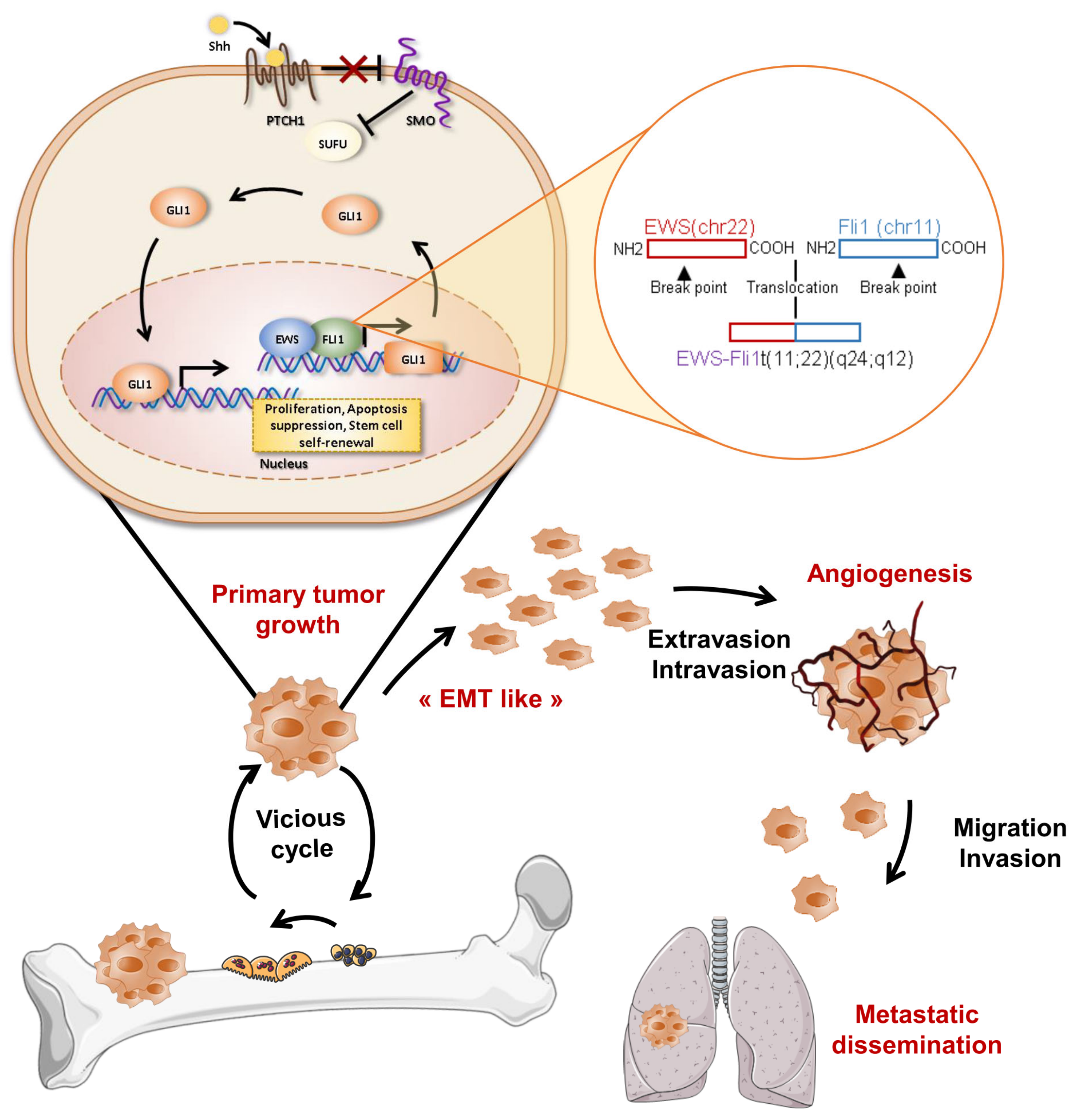
5. SHH Signaling in OS et ES
6. Conclusions and Clinical Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- ESMO/European Sarcoma Network Working Group Bone sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, 113–123. [CrossRef] [PubMed]
- Spina, V.; Montanari, N.; Romagnoli, R. Malignant tumors of the osteogenic matrix. Eur. J. Radiol. 1998, 27, S98–S109. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D.M. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Ritter, J.; Bielack, S.S. Osteosarcoma. Ann. Oncol. 2010, 21, 320–325. [Google Scholar] [CrossRef]
- Bousquet, M.; Noirot, C.; Accadbled, F.; De Gauzy, J.S.; Castex, M.P.; Brousset, P.; Gomez-Brouchet, A. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann. Oncol. 2016, 27, 738–744. [Google Scholar] [CrossRef]
- Eccles, S.A.; Welch, D.R. Metastasis: recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef] [Green Version]
- Dockhorn-Dworniczak, B.; Schäfer, K.L.; Dantcheva, R.; Blasius, S.; Winkelmann, W.; Strehl, S.; Burdach, S.; van Valen, F.; Jürgens, H.; Böcker, W. Diagnostic value of the molecular genetic detection of the t(11;22) translocation in Ewing’s tumours. Virchows Arch. 1994, 425, 107–112. [Google Scholar] [CrossRef]
- Heare, T.; Hensley, M.A.; Dell’Orfano, S. Bone tumors: osteosarcoma and Ewing’s sarcoma. Curr. Opin. Pediatr. 2009, 21, 365–372. [Google Scholar] [CrossRef]
- Patricio, M.B.; Vilhena, M.; Neves, M.; Raposo, S.; Catita, J.; De Sousa, V.; Martins, A.G. Ewing’s sarcoma in children: twenty-five years of experience at the Instituto Portugês de Oncologia de Francisco Gentil (I.P.O.F.G.). J. Surg. Oncol. 1991, 47, 37–40. [Google Scholar] [CrossRef]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.H.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; McManus, M.M.; Hughes, T.P. Understanding the Biology of Bone Sarcoma from Early Initiating Events through Late Events in Metastasis and Disease Progression. Front. Oncol. 2013, 3, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortus, J.R.; Zhang, Y.; Hughes, T.P. Developmental Pathways Hijacked by Osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 93–118. [Google Scholar] [PubMed]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Ogaki, M.; Yanae, M.; Nishida, S. Reduction of metastasis, cell invasion, and adhesion in mouse osteosarcoma by YM529/ONO-5920-induced blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathway. Toxicol. Appl. Pharmacol. 2012, 259, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-J.; Lee, T.-S.; Park, J.-B.; Park, K.-K.; Choe, J.-Y.; Sin, D.-I.; Park, Y.-Y.; Moon, Y.S.; Lee, K.-G.; Yeo, J.-H.; et al. Disulfiram suppresses invasive ability of osteosarcoma cells via the inhibition of MMP-2 and MMP-9 expression. J. Biochem. Mol. Biol. 2007, 40, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.E.; Bekarev, M.; Kim, M.; Lin, J.; Piperdi, S.; Gorlick, R.; Geller, D.S. Cell surface receptor expression patterns in osteosarcoma. Cancer 2011, 118, 740–749. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Davis, L.E.; Bolejack, V.; Ryan, C.W.; Ganjoo, K.N.; Loggers, E.T.; Chawla, S.; Agulnik, M.; Livingston, M.B.; Reed, D.; Keedy, V.; et al. Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J. Clin. Oncol. 2019, 37, 1424–1431. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [Green Version]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Jeong, J.; McMahon, A.P. Cholesterol modification of Hedgehog family proteins. J. Clin. Investig. 2002, 110, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Wendler, F.; Franch-Marro, X.; Vincent, J.-P. How does cholesterol affect the way Hedgehog works? Development 2006, 133, 3055–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassov, A.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Parchure, A.; Vyas, N.; Mayor, S. Wnt and Hedgehog: Secretion of Lipid-Modified Morphogens. Trends Cell Boil. 2017, 28, 157–170. [Google Scholar] [CrossRef]
- Marigo, V.; Davey, R.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that Patched is the Hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef]
- Incardona, J.P.; Lee, J.H.; Robertson, C.P.; Enga, K.; Kapur, R.P.; Roelink, H. Receptor-mediated endocytosis of soluble and membrane-tethered Sonic hedgehog by Patched. Proc. Natl. Acad. Sci. USA 2000, 97, 12044–12049. [Google Scholar] [CrossRef] [Green Version]
- Pham, A.; Therond, P.; Alves, G.; Tournier, F.B.; Busson, D.; Lamour-Isnard, C.; Bouchon, B.L.; Preat, T.; Tricoire, H. The Suppressor of Fused Gene Encodes a Novel Pest Protein Involved in Drosophila Segment Polarity Establishment. Genetics 1995, 140, 587–598. [Google Scholar]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.-C.; et al. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Boil. 2017, 37, e00421-16. [Google Scholar] [CrossRef] [Green Version]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Boil. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavletich, N.; Pabo, C. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell. Boil. 1988, 8, 3104–3113. [Google Scholar] [CrossRef]
- Hui, C.-C.; Slusarski, D.; Platt, K.A.; Holmgren, R.; Joyner, A.L. Expression of Three Mouse Homologs of the Drosophila Segment Polarity Gene cubitus interruptus, Gli, Gli-2, and Gli-3, in Ectoderm- and Mesoderm-Derived Tissues Suggests Multiple Roles during Postimplantation Development. Dev. Boil. 1994, 162, 402–413. [Google Scholar] [CrossRef]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Leprieur, E.G.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective Modulation of Hedgehog/GLI Target Gene Expression by Epidermal Growth Factor Signaling in Human Keratinocytes†. Mol. Cell. Boil. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [Green Version]
- Alexaki, V.-I.; Javelaud, D.; Van Kempen, L.C.L.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Juàrez, P.; Goydos, J.S.; Fournier, P.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; André, J.; Verrecchia, F.; Mauviel, A. Cloning of the Human GLI2 Promoter. J. Boil. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Üren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Boil. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, B.; Huang, R.; Wilting, J. The development of the avian vertebral column. Brain Struct. Funct. 2000, 202, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, H.; Mizutani-Koseki, S.; Koseki, H. Three developmental compartments involved in rib formation. Int. J. Dev. Boil. 2005, 49, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, J.M.; Teillet, M.-A.; Le Douarin, N.M. An early role for Sonic hedgehog from foregut endoderm in jaw development: Ensuring neural crest cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 11607–11612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calloni, G.W.; Glavieux-Pardanaud, C.; Le Douarin, N.M.; Dupin, E. Sonic Hedgehog promotes the development of multipotent neural crest progenitors endowed with both mesenchymal and neural potentials. Proc. Natl. Acad. Sci. USA 2007, 104, 19879–19884. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.L.; Di Franco, M.; Eichers, E.; May-Simera, H.; Garcia, M.; Yan, J.; Quinlan, R.; Justice, M.J.; Hennekam, R.C.; Briscoe, J.; et al. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6714–6719. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, R.; Kawakami, H.; Taketo, M.M.; Evans, S.M.; Wada, N.; Petryk, A.; Kawakami, Y. Distinct populations within Isl1 lineages contribute to appendicular and facial skeletogenesis through the β-catenin pathway. Dev. Boil. 2014, 387, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Mao, J.; Tenzen, T.; Kottmann, A.; McMahon, A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genome Res. 2004, 18, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Eberhart, J.K. Early Hedgehog signaling from neural to oral epithelium organizes anterior craniofacial development. Development 2006, 133, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate SHH signaling during mouse development. Genome Res. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orioli, I.M.; Vieira, A.R.; Castilla, E.E.; Ming, J.E.; Muenke, M. Mutational analysis of theSonic Hedgehoggene in 220 newborns with oral clefts in a South American (ECLAMC) population†. Am. J. Med Genet. 2002, 108, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Lettice, L.A.; Heaney, S.J.; Purdie, L.A.; Li, L.; De Beer, P.; Oostra, B.A.; Goode, D.; Elgar, G.; Hill, R.E.; De Graaff, E. A long-range SHH enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003, 12, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.; De La Cruz, J.; Lewis, R.A.; Karkera, J.; Manligas, G.S.; Roessler, E.; Muenke, M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med Genet. 2002, 116, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.K. Solitary median maxillary central incisor (SMMCI) syndrome. Orphanet J. Rare Dis. 2006, 1, 12. [Google Scholar] [CrossRef]
- Aoto, K.; Shikata, Y.; Imai, H.; Matsumaru, D.; Tokunaga, T.; Shioda, S.; Yamada, G.; Motoyama, J. Mouse SHH is required for prechordal plate maintenance during brain and craniofacial morphogenesis. Dev. Boil. 2009, 327, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Balczerski, B.; Zakaria, S.; Tucker, A.S.; Borycki, A.; Koyama, E.; Pacifici, M.; Francis-West, P. Distinct spatiotemporal roles of hedgehog signaling during chick and mouse cranial base and axial skeleton development. Dev. Boil. 2012, 371, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Martí, E.; Takada, R.; A Bumcrot, D.; Sasaki, H.; McMahon, A.P. Distribution of Sonic hedgehog peptides in the developing chick and mouse embryo. Development 1995, 121, 2537–2547. [Google Scholar]
- Poelmans, S.; Kawamoto, T.; Cristofoli, F.; Politis, C.; Vermeesch, J.; Bailleul-Forestier, I.; Hens, G.; Devriendt, K.; Verdonck, A.; Carels, C. Genotypic and phenotypic variation in six patients with solitary median maxillary central incisor syndrome. Am. J. Med Genet. Part A 2015, 167, 2451–2458. [Google Scholar] [CrossRef]
- Everson, J.; Fink, D.M.; Yoon, J.W.; Leslie, E.J.; Kietzman, H.W.; Ansen-Wilson, L.J.; Chung, H.M.; Walterhouse, D.O.; Marazita, M.L.; Lipinski, R. Sonic hedgehog regulation of Foxf2 promotes cranial neural crest mesenchyme proliferation and is disrupted in cleft lip morphogenesis. Development 2017, 144, 2082–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, N.; Sidik, A.; Bertrand, J.; Eberhart, J.K. An Fgf-SHH signaling hierarchy regulates early specification of the zebrafish skull. Dev. Boil. 2016, 415, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Ramalho-Santos, M.; McMahon, A.P. Smoothened Mutants Reveal Redundant Roles for SHH and IHH Signaling Including Regulation of L/R Asymmetry by the Mouse Node. Cell 2001, 105, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Zahid, S.; Shilts, M.H.; Weaver, S.J.; Leskowitz, R.M.; Habbsa, S.; Aronowitz, D.; Rokins, K.P.; Chang, Y.; Pinnella, Z.; et al. Hoxa-5 acts in segmented somites to regulate cervical vertebral morphology. Mech. Dev. 2013, 130, 226–240. [Google Scholar] [CrossRef]
- Zuniga, A.; Haramis, A.-P.G.; McMahon, A.P.; Zeller, R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature 1999, 401, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, S.; Boglev, Y.; Owens, H.; Goldie, S.J. The Role of Sonic Hedgehog in Craniofacial Patterning, Morphogenesis and Cranial Neural Crest Survival. J. Dev. Boil. 2016, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Xavier, G.M.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M.T. Hedgehog receptor function during craniofacial development. Dev. Boil. 2016, 415, 198–215. [Google Scholar] [CrossRef] [Green Version]
- Dassule, H.R.; Lewis, P.; Bei, M.; Maas, R.L.; McMahon, A.P. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 2000, 127, 4775–4785. [Google Scholar]
- Drossopoulou, G.; E Lewis, K.; Sanz-Ezquerro, J.J.; Nikbakht, N.; McMahon, A.P.; Hofmann, C.; Tickle, C. A model for anteroposterior patterning of the vertebrate limb based on sequential long- and short-range SHH signaling and Bmp signaling. Development 2000, 127, 1337–1348. [Google Scholar]
- Firulli, B.A.; Fuchs, R.K.; Vincentz, J.W.; Clouthier, D.E.; Firulli, A.B. Hand1 phosphoregulation within the distal arch neural crest is essential for craniofacial morphogenesis. Development 2014, 141, 3050–3061. [Google Scholar] [CrossRef] [Green Version]
- Melnick, M.; Witcher, D.P.B., Jr.; Carlsson, P.; Jaskoll, T. Meckel’s Cartilage Differentiation Is Dependent on Hedgehog Signaling. Cells Tissues Organs 2005, 179, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Dassule, H.R.; McMahon, A.P. Analysis of Epithelial–Mesenchymal Interactions in the Initial Morphogenesis of the Mammalian Tooth. Dev. Boil. 1998, 202, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritli-Linde, A. SHH signaling within the dental epithelium is necessary for cell proliferation, growth and polarization. Development 2002, 129, 5323–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, L.A.; Richieri-Costa, A. Single median maxillary central incisor, hypophyseal tumor, andSHH mutation. Am. J. Med Genet. Part A 2005, 136, 346–347. [Google Scholar] [CrossRef]
- Cobourne, M.T.; Sharpe, P. Making up the numbers: The molecular control of mammalian dental formula. Semin. Cell Dev. Boil. 2010, 21, 314–324. [Google Scholar] [CrossRef]
- Paiva, K.; Silva-Valenzuela, M.D.G.; Massironi, S.M.G.; Ko, G.M.; Siqueira, F.M.; Nunes, F.D. Differential SHH, Bmp and Wnt gene expressions during craniofacial development in mice. Acta Histochem. 2010, 112, 508–517. [Google Scholar] [CrossRef]
- Hovorakova, M.; Smrckova, L.; Lesot, H.; Lochovska, K.; Peterka, M.; Peterková, R. Sequential SHH expression in the development of the mouse upper functional incisor. J. Exp. Zool. Part B: Mol. Dev. Evol. 2013, 320, 455–464. [Google Scholar]
- Abramyan, J.; Richman, J.M. Craniofacial development: discoveries made in the chicken embryo. Int. J. Dev. Boil. 2018, 62, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Dutka, T.; Devenney, B.M.; Kawasaki, K.; Reeves, R.H.; Richtsmeier, J.T. Acute upregulation of hedgehog signaling in mice causes differential effects on cranial morphology. Dis. Model. Mech. 2014, 8, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Kaucka, M.; Petersen, J.; Tesarova, M.; Szarowska, B.; Kastriti, M.E.; Xie, M.; Kicheva, A.; Annusver, K.; Kasper, M.; Symmons, O.; et al. Signals from the brain and olfactory epithelium control shaping of the mammalian nasal capsule cartilage. eLife 2018, 7, e34465. [Google Scholar] [CrossRef]
- Hu, D.; Marcucio, R. Unique organization of the frontonasal ectodermal zone in birds and mammals. Dev. Boil. 2008, 325, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcucio, R.; Cordero, D.R.; Hu, D.; Arioka, M. Molecular interactions coordinating the development of the forebrain and face. Dev. Boil. 2005, 284, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabin, C.J.; McMahon, A.P. DEVELOPMENTAL BIOLOGY: Grasping Limb Patterning. Science 2008, 321, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Vokes, S.A.; Ji, H.; Wong, W.H.; McMahon, A.P. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genome Res. 2008, 22, 2651–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz, P.J.; Harfe, B.D.; McMahon, A.P.; Tabin, C.J. The Limb Bud SHH-Fgf Feedback Loop Is Terminated by Expansion of Former ZPA Cells. Science 2004, 305, 396–399. [Google Scholar] [CrossRef]
- A Parr, B.; McMahon, A.P. Dorsalizing signal Wnt-7a required for normal polarity of D–V and A–P axes of mouse limb. Nature 1995, 374, 350–353. [Google Scholar] [CrossRef]
- Harfe, B.D.; Scherz, P.J.; Nissim, S.; Tian, H.; McMahon, A.P.; Tabin, C.J. Evidence for an Expansion-Based Temporal SHH Gradient in Specifying Vertebrate Digit Identities. Cell 2004, 118, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, R.; Kawakami, H.; Wong, J.; Oishi, I.; Nishinakamura, R.; Kawakami, Y. Sall4-Gli3 system in early limb progenitors is essential for the development of limb skeletal elements. Proc. Natl. Acad. Sci. USA 2015, 112, 5075–5080. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M. Fins into limbs: Autopod acquisition and anterior elements reduction by modifying gene networks involving 5’Hox, Gli3, and SHH. Dev. Boil. 2016, 413, 1–7. [Google Scholar] [CrossRef]
- Reinhardt, R.; Gullotta, F.; Nusspaumer, G.; Ünal, E.; Ivanek, R.; Zuniga, A.; Zeller, R. Molecular signatures identify immature mesenchymal progenitors in early mouse limb buds that respond differentially to morphogen signaling. Development 2019, 146, dev173328. [Google Scholar] [CrossRef] [Green Version]
- Litingtung, Y.; Dahn, R.D.; Li, Y.; Fallon, J.F.; Chiang, C. SHH and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 2002, 418, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, T.B.; Kochhar, D.M. The Hemimelic extra toes mouse mutant: Historical perspective on unraveling mechanisms of dysmorphogenesis. Birth Defects Res. Part C: Embryo Today: Rev. 2010, 90, 155–162. [Google Scholar] [CrossRef]
- Lopez-Rios, J. The many lives of SHH in limb development and evolution. Semin. Cell Dev. Boil. 2016, 49, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Niedermaier, M.; Schwabe, G.C.; Fees, S.; Helmrich, A.; Brieske, N.; Seemann, P.; Hecht, J.; Seitz, V.; Stricker, S.; Leschik, G.; et al. An inversion involving the mouse SHH locus results in brachydactyly through dysregulation of SHH expression. J. Clin. Investig. 2005, 115, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, D.; Pawlik, B.; Li, Y.; Akarsu, N.A.; Caliebe, A.; May, K.J.; Schweiger, B.; Vargas, F.R.; Balci, S.; Gillessen-Kaesbach, G.; et al. A specific mutation in the distant sonic hedgehog (SHH)cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum. Mutat. 2010, 31, 81–89. [Google Scholar] [CrossRef]
- Al-Qattan, M.M.; Al Abdulkareem, I.; Alhaidan, Y.; Al Balwi, M.A. A novel mutation in theSHHlong-range regulator (ZRS) is associated with preaxial polydactyly, triphalangeal thumb, and severe radial ray deficiency. Am. J. Med Genet. Part A 2012, 158, 2610–2615. [Google Scholar] [CrossRef]
- Gurnett, C.; Bowcock, A.; Dietz, F.R.; Morcuende, J.A.; Murray, J.C.; Dobbs, M.B. Two novel point mutations in the long-range SHH enhancer in three families with triphalangeal thumb and preaxial polydactyly. Am. J. Med Genet. Part A 2007, 143, 27–32. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; A Beachy, P. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nat. 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Aguinaga, M.; Zenteno, J.C.; Moran, V.; Pérez-Cano, H. Sonic hedgehog mutation analysis in patients with VACTERL association. Am. J. Med Genet. Part A 2010, 152, 781–783. [Google Scholar] [CrossRef]
- Choi, K.-S.; Lee, C.; Harfe, B.D. Sonic hedgehog in the notochord is sufficient for patterning of the intervertebral discs. Mech. Dev. 2012, 129, 255–262. [Google Scholar] [CrossRef]
- Lubinsky, M. Sonic Hedgehog, VACTERL, and Fanconi anemia: Pathogenetic connections and therapeutic implications. Am. J. Med Genet. Part A 2015, 167, 2594–2598. [Google Scholar] [CrossRef] [PubMed]
- Orchard, P.; White, J.S.; E Thomas, P.; Mychalowych, A.; Kiseleva, A.; Hensley, J.; Allen, B.; Parker, S.C.; E Keegan, C. Genome-wide chromatin accessibility and transcriptome profiling show minimal epigenome changes and coordinated transcriptional dysregulation of hedgehog signaling in Danforth’s short tail mice. Hum. Mol. Genet. 2018, 28, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Jemtland, R.; Divieti, P.; Lee, K.; Segre, G.V. Hedgehog promotes primary osteoblast differentiation and increases PTHrP mRNA expression and iPTHrP secretion. Bone 2003, 32, 611–620. [Google Scholar] [CrossRef]
- Tsiairis, C.; McMahon, A.P. Disp1 regulates growth of mammalian long bones through the control of IHH distribution. Dev. Boil. 2008, 317, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-F.; Serra, R. Ift88 regulates Hedgehog signaling, Sfrp5 expression, and β-catenin activity in post-natal growth plate. J. Orthop. Res. 2012, 31, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; Kito, A.; Itoh, S.; Naruse, H.; Fujikawa, J.; Sadek, K.M.; Akiyama, S.; Yamashiro, T.; Wakisaka, S.; Abe, M. Kruppel-Like Factor 4 represses osteoblast differentiation via ciliary Hedgehog signaling. Exp. Cell Res. 2018, 371, 417–425. [Google Scholar] [CrossRef]
- Hojo, H.; Ohba, S.; Taniguchi, K.; Shirai, M.; Yano, F.; Saito, T.; Ikeda, T.; Nakajima, K.; Komiyama, Y.; Nakagata, N.; et al. Hedgehog-Gli Activators Direct Osteo-chondrogenic Function of Bone Morphogenetic Protein toward Osteogenesis in the Perichondrium*. J. Boil. Chem. 2013, 288, 9924–9932. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-H.; White, K.A.; Somatilaka, B.N.; Shelton, J.M.; Richardson, J.A.; Mukhopadhyay, S. The G protein-coupled receptor Gpr161 regulates forelimb formation, limb patterning and skeletal morphogenesis in a primary cilium-dependent manner. Development 2018, 145, dev154054. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, M.; Solomon, J.; Ghali, B.; Van Der Meulen, M.C.; Crystal, R.G.; Hidaka, C. Transient Overexpression of Sonic Hedgehog Alters the Architecture and Mechanical Properties of Trabecular Bone. J. Bone Miner. Res. 2009, 24, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Iwakura, T.; Inui, A.; Reddi, A.H. Stimulation of superficial zone protein accumulation by hedgehog and Wnt signaling in surface zone bovine articular chondrocytes. Arthritis Rheum. 2013, 65, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, B.E.; Henner, A.; Stewart, S.; Stankunas, K. SHH promotes direct interactions between epidermal cells and osteoblast progenitors to shape regenerated zebrafish bone. Development 2017, 144, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Shimo, T.; Kurio, N.; Okui, T.; Obata, K.; Masui, M.; Pang, P.; Horikiri, Y.; Sasaki, A. Expression and Role of Sonic Hedgehog in the Process of Fracture Healing with Aging. In Vivo 2016, 30, 99–105. [Google Scholar]
- Lin, L.; Shen, Q.; Xue, T.; Duan, X.; Fu, X.; Yu, C. Sonic Hedgehog Improves Redifferentiation of Dedifferentiated Chondrocytes for Articular Cartilage Repair. PLoS ONE 2014, 9, e88550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Hui, C.-C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, L.; Beachy, P.A. The Hedgehog response network: sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimkus, T.K.; Carpenter, R.; Qasem, S.A.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Di Magliano, M.P.; Hebrok, M. Hedgehog signaling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.-W. Hedgehog pathway and GLI1 isoforms in human cancer. Discov. Med. 2012, 13, 105–113. [Google Scholar]
- Villavicencio, E.H.; Walterhouse, D.O.; Iannaccone, P.M. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000, 67, 1047–1054. [Google Scholar] [CrossRef]
- Ng, J.M.Y.; Curran, T. The Hedgehog’s tale: developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.-C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human Homolog of patched, a Candidate Gene for the Basal Cell Nevus Syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vořechovský, I.; Holmberg, E.; Undèn, A.B.; Gillies, S.; et al. Mutations of the Human Homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma Syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Oro, A.E. Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.M.; Douglass, E.C.; Peiper, S.C.; Houghton, P.J.; Look, A.T. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989, 49, 5407–5413. [Google Scholar] [PubMed]
- Hirotsu, M.; Setoguchi, T.; Sasaki, H.; Matsunoshita, Y.; Gao, H.; Nagao, H.; Kunigou, O.; Komiya, S. Smoothened as a new therapeutic target for human osteosarcoma. Mol. Cancer 2010, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.W.; Pinnaduwage, D.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L. Aberrant Hedgehog Signaling and Clinical Outcome in Osteosarcoma. Sarcoma 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Liu, X.; Choy, E.; Mankin, H.; Hornicek, F.J.; Duan, Z. Targeting hedgehog-GLI-2 pathway in osteosarcoma. J. Orthop. Res. 2012, 31, 502–509. [Google Scholar] [CrossRef]
- Nagao, H.; Hirotsu, M.; Ishidou, Y.; Nagano, S.; Takizawa, T.; Nakashima, K.; Setoguchi, T.; Ijiri, K.; Yamamoto, T.; Komiya, S. Role of GLI2 in the growth of human osteosarcoma†. J. Pathol. 2011, 224, 169–179. [Google Scholar] [CrossRef]
- Lo, W.W.; Wunder, J.S.; Dickson, B.C.; Campbell, V.; McGovern, K.; Alman, B.; Andrulis, I.L. Involvement and targeted intervention of dysregulated Hedgehog signaling in osteosarcoma. Cancer 2013, 120, 537–547. [Google Scholar] [CrossRef]
- Nakamura, S.; Nagano, S.; Nagao, H.; Ishidou, Y.; Yokouchi, M.; Abematsu, M.; Yamamoto, T.; Komiya, S.; Setoguchi, T. Arsenic Trioxide Prevents Osteosarcoma Growth by Inhibition of GLI Transcription via DNA Damage Accumulation. PLoS ONE 2013, 8, e69466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2010, 121, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Lin, C.-P.; Hsu, M.-L.; Shieh, H.-R.; Chao, N.K.; Chao, K.C. Sonic Hedgehog Signaling Protects Human Hepatocellular Carcinoma Cells Against Ionizing Radiation in an Autocrine Manner. Int. J. Radiat. Oncol. 2011, 80, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Wang, Y.; Wu, Q.; Hao, D.; Li, D. Emodin Impairs Radioresistance of Human Osteosarcoma Cells by Suppressing Sonic Hedgehog Signaling. Med. Sci. Monit. 2017, 23, 5767–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, W.; Li, D.; Wang, Y.; Wu, Q.; Hao, D. Activation of Sonic Hedgehog Signaling Is Associated with Human Osteosarcoma Cells Radioresistance Characterized by Increased Proliferation, Migration, and Invasion. Med. Sci. Monit. 2018, 24, 3764–3771. [Google Scholar] [CrossRef]
- Qi, J.; Zhou, Y.; Jiao, Z.; Wang, X.; Zhao, Y.; Li, Y.; Chen, H.; Yang, L.; Zhu, H.; Li, Y. Exosomes Derived from Human Bone Marrow Mesenchymal Stem Cells Promote Tumor Growth Through Hedgehog Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2242–2254. [Google Scholar] [CrossRef]
- Wang, N.; Li, P.; Liu, W.; Lu, Z.; Feng, J.; Zeng, X.; Yang, J.; Zhao, W. miR-141-3p suppresses proliferation and promotes apoptosis by targeting GLI2 in osteosarcoma cells. Oncol. Rep. 2017, 39, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, K.-L.; Eisenacher, M.; Braun, Y.; Brachwitz, K.; Wai, D.H.; Dirksen, U.; Lanvers-Kaminsky, C.; Juergens, H.; Herrero, D.; Stegmaier, S.; et al. Microarray analysis of Ewing’s sarcoma family of tumours reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur. J. Cancer 2008, 44, 699–709. [Google Scholar] [CrossRef]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.-G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 Is a Central Mediator of EWS/FLI1 Signaling in Ewing Tumors. PLoS ONE 2009, 4, 7608. [Google Scholar] [CrossRef]
- Peukert, S.; Miller-Moslin, K. Small-Molecule Inhibitors of the Hedgehog Signaling Pathway as Cancer Therapeutics. ChemMedChem 2010, 5, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Mahindroo, N.; Punchihewa, C.; Fujii, N. Hedgehog-Gli Signaling Pathway Inhibitors as Anticancer Agents. J. Med. Chem. 2009, 52, 3829–3845. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genome Res. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Italiano, A.; Mir, O.; Cioffi, A.; Palmerini, E.; Piperno-Neumann, S.; Perrin, C.; Chaigneau, L.; Penel, N.; Duffaud, F.; Kurtz, J.E.; et al. Advanced chondrosarcomas: role of chemotherapy and survival. Ann. Oncol. 2013, 24, 2916–2922. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lézot, F.; Corre, I.; Morice, S.; Rédini, F.; Verrecchia, F. SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression. Cells 2020, 9, 536. https://doi.org/10.3390/cells9030536
Lézot F, Corre I, Morice S, Rédini F, Verrecchia F. SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression. Cells. 2020; 9(3):536. https://doi.org/10.3390/cells9030536
Chicago/Turabian StyleLézot, Frédéric, Isabelle Corre, Sarah Morice, Françoise Rédini, and Franck Verrecchia. 2020. "SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression" Cells 9, no. 3: 536. https://doi.org/10.3390/cells9030536
APA StyleLézot, F., Corre, I., Morice, S., Rédini, F., & Verrecchia, F. (2020). SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression. Cells, 9(3), 536. https://doi.org/10.3390/cells9030536