Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms



Abstract
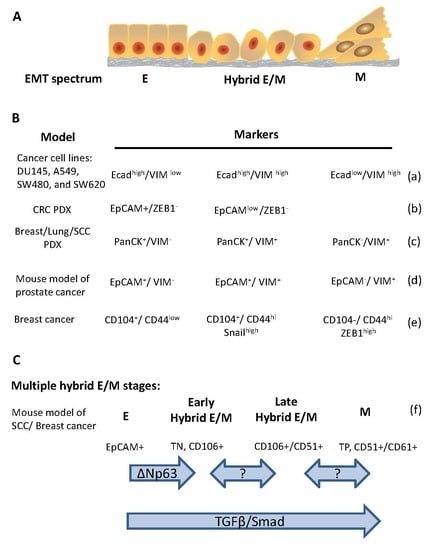
:
{kind=link}
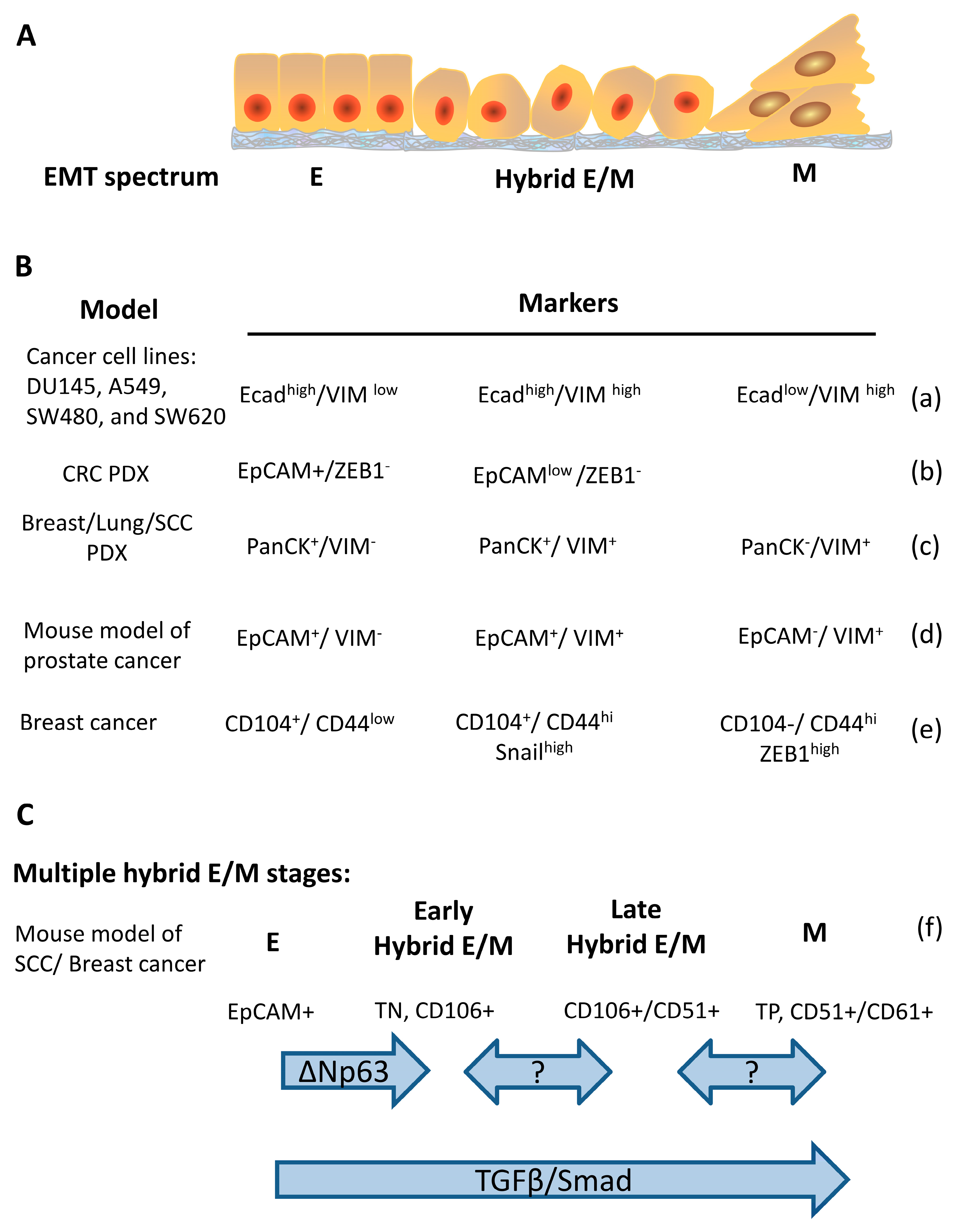{kind=link}
1. Introduction
2. Evidence of Hybrid E/M in Cancers
3. The Regulation of Hybrid E/M in Cancer Cells
3.1. Conventional EMT Inducers
3.2. Transcriptional Regulation of Hybrid E/M
3.3. Epigenetic Regulation of Hybrid E/M
4. Hybrid E/M and Cancer Stemness
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Greenburg, G.; Hay, E.D. Cytoskeleton and thyroglobulin expression change during transformation of thyroid epithelium to mesenchyme–like cells. Development 1988, 102, 605–622. [Google Scholar] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, E.D. An overview of epithelio–mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial–mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer. 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal–epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial–mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival outcomes in cancer patients predicted by a partial EMT gene expression scoring metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [Green Version]
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach–Carson, M.C.; Levine, H. Tumor budding: The name is EMT. partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression–free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Mostert, B.; Sleijfer, S.; Foekens, J.A.; Gratama, J.W. Circulating tumor cells (CTCs): Detection methods and their clinical relevance in breast cancer. Cancer Treat. Rev. 2009, 35, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia–Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef]
- Friedl, P.; Mayor, R. Tuning collective cell migration by cell–cell junction regulation. Cold Spring Harb. Perspect. Biol. 2017, 9, a029199. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF–1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2–mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal–like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.A.; Kirschmann, D.A.; Cerhan, J.R.; Folberg, R.; Seftor, E.A.; Sellers, T.A.; Hendrix, M.J. Association between keratin and vimentin expression, malignant phenotype, and survival in postmenopausal breast cancer patients. Clin. Cancer Res. 1999, 5, 2698–2703. [Google Scholar] [PubMed]
- Kolijn, K.; Verhoef, E.I.; van Leenders, G.J. Morphological and immunohistochemical identification of epithelial–to–mesenchymal transition in clinical prostate cancer. Oncotarget 2015, 6, 24488–24498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharias, M.; Brcic, L.; Eidenhammer, S.; Popper, H. Bulk tumour cell migration in lung carcinomas might be more common than epithelial–mesenchymal transition and be differently regulated. BMC Cancer 2018, 18, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, N.; Hoshida, Y.; Aguirre–Ghiso, J.A. Origin and interpretation of cancer transcriptome profiling: The essential role of the stroma in determining prognosis and drug resistance. EMBO Mol. Med. 2015, 7, 1385–1387. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single–cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Mizukoshi, K.; Okazawa, Y.; Haeno, H.; Koyama, Y.; Sulidan, K.; Komiyama, H.; Saeki, H.; Ohtsuji, N.; Ito, Y.; Kojima, Y.; et al. Metastatic seeding of human colon cancer cell clusters expressing the hybrid epithelial/mesenchymal state. Int. J. Cancer. 2019. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Chao, C.; Widen, S.G.; Wood, T.G.; Zatarain, J.R.; Johnson, P.; Gajjar, A.; Gomez, G.; Qiu, S.; Thompson, J.; Spratt, H.; et al. Patient–derived xenografts from colorectal carcinoma: A temporal and hierarchical study of murine stromal cell replacement. Anticancer Res. 2017, 37, 3405–3412. [Google Scholar]
- Kopetz, S.; Lemos, R.; Powis, G. The promise of patient–derived xenografts: The best laid plans of mice and men. Clin. Cancer Res. 2012, 18, 5160–5162. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi–Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial–mesenchymal plasticity and suppresses metastatic, castration–resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Trevor, K.T. Experimental co–expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am. J. Pathol. 1997, 150, 483–495. [Google Scholar] [PubMed]
- Bronsert, P.; Enderle–Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three–dimensional study of the human cancer–host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin–beta4 identifies cancer stem cell–enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.Y.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.; Chung, V.Y.; Chu, Y.S.; Matsumura, N.; Lai, H.C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis–resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e–cadherin restoration by a src–kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem–cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Sampson, V.B.; David, J.M.; Puig, I.; Patil, P.U.; de Herreros, A.G.; Thomas, G.V.; Rajasekaran, A.K. Wilms’ tumor protein induces an epithelial–mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS ONE 2014, 9, e102041. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial–mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial–mesenchymal and mesenchymal–epithelial transitions by microRNAs. Curr. Opin. Cell Biol. 2013, 25, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1–mediated transcriptional repression during epithelial–mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E–cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E–cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez–Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E–cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Diaz, V.M.; Franci, C.; Gutierrez, A.; Dave, N.; Escriva, M.; Hernandez–Munoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E–cadherin repression by the Snail1 transcription factor. Mol. Cell Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine–specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium–mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116 (Pt 10), 1959–1967. [Google Scholar] [CrossRef] [Green Version]
- Stanisavljevic, J.; Porta–de–la–Riva, M.; Batlle, R.; de Herreros, A.G.; Baulida, J. The p65 subunit of NF–kappaB and PARP1 assist Snail1 in activating fibronectin transcription. J. Cell Sci. 2011, 124 (Pt 24), 4161–4171. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.S.; Lan, H.Y.; Huang, C.H.; Tai, S.K.; Chang, S.Y.; Tsai, T.L.; Chang, C.C.; Tzeng, C.H.; Wu, K.J.; Kao, J.Y.; et al. Regulation of excision repair cross–complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin. Cancer Res. 2010, 16, 4561–4571. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.L.; Yang, M.H.; Tsai, M.L.; Lan, H.Y.; Su, S.H.; Chang, S.C.; Teng, H.W.; Yang, S.H.; Lan, Y.T.; Chiou, S.H.; et al. SNAIL regulates interleukin–8 expression, stem cell–like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Spoelstra, N.S.; Manning, N.G.; Higashi, Y.; Darling, D.; Singh, M.; Shroyer, K.R.; Broaddus, R.R.; Horwitz, K.B.; Richer, J.K. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006, 66, 3893–3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E–cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, C.; Garcia, J.M.; Garcia, V.; Silva, J.; Dominguez, G.; Rodriguez, R.; Maximiano, C.; Garcia de Herreros, A.; Munoz, A.; Bonilla, F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E–cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer. 2006, 119, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Revisiting epithelial–mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804. [Google Scholar] [CrossRef]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2562. [Google Scholar] [CrossRef] [Green Version]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal–Ponce, A.; Nie, Q.; Dai, X. An Ovol2–Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi–step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef]
- Kitazawa, K.; Hikichi, T.; Nakamura, T.; Mitsunaga, K.; Tanaka, A.; Nakamura, M.; Yamakawa, T.; Furukawa, S.; Takasaka, M.; Goshima, N.; et al. OVOL2 maintains the transcriptional program of human corneal epithelium by suppressing epithelial–to–mesenchymal transition. Cell Rep. 2016, 15, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Villarreal–Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew–Goodall, Y.; Goodall, G.J. The miR–200 family and miR–205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR–200 family inhibits epithelial–mesenchymal transition and cancer cell migration by direct targeting of E–cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR–200 family determines the epithelial phenotype of cancer cells by targeting the E–cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double–negative feedback loop between ZEB1–SIP1 and the microRNA–200 family regulates epithelial–mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF–beta/ZEB/miR–200 signaling network regulates establishment and maintenance of epithelial–mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1–induced epithelial–mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lee, D.K.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell–specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial–to–mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK–3beta–mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Yoon, S.O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase–3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial–mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt–Axin2–GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.; Balaji, K.C. Protein kinase D1 suppresses epithelial–to–mesenchymal transition through phosphorylation of snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Rayala, S.; Nguyen, D.; Vadlamudi, R.K.; Chen, S.; Kumar, R. Pak1 phosphorylation of snail, a master regulator of epithelial–to–mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 2005, 65, 3179–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Rodriguez–Aznar, E.; Yabuta, N.; Owen, R.J.; Mingot, J.M.; Nojima, H.; Nieto, M.A.; Longmore, G.D. Lats2 kinase potentiates Snail1 activity by promoting nuclear retention upon phosphorylation. EMBO J. 2012, 31, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, D.S.; Wang, H.J.; Tai, S.K.; Chou, C.H.; Hsieh, C.H.; Chiu, P.H.; Chen, N.J.; Yang, M.H. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell 2014, 26, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCart Reed, A.E.; Kutasovic, J.R.; Vargas, A.C.; Jayanthan, J.; Al–Murrani, A.; Reid, L.E.; Chambers, R.; Da Silva, L.; Melville, L.; Evans, E.; et al. An epithelial to mesenchymal transition programme does not usually drive the phenotype of invasive lobular carcinomas. J. Pathol. 2016, 238, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E–cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [Green Version]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail–induced claudin–11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial–mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Meca-Cortes, O.; Mateo, F.; Martinez de Paz, A.; Rubio, N.; Arnal-Estape, A.; Ell, B.J.; Bermudo, R.; Diaz, A.; Guerra-Rebollo, M.; et al. Epithelial–mesenchymal transition can suppress major attributes of human epithelial tumor–initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [Green Version]
- Fustaino, V.; Presutti, D.; Colombo, T.; Cardinali, B.; Papoff, G.; Brandi, R.; Bertolazzi, P.; Felici, G.; Ruberti, G. Characterization of epithelial–mesenchymal transition intermediate/hybrid phenotypes associated to resistance to EGFR inhibitors in non–small cell lung cancer cell lines. Oncotarget 2017, 8, 103340–103363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy–induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiscox, S.; Jiang, W.G.; Obermeier, K.; Taylor, K.; Morgan, L.; Burmi, R.; Barrow, D.; Nicholson, R.I. Tamoxifen resistance in MCF7 cells promotes EMT–like behaviour and involves modulation of beta–catenin phosphorylation. Int J. Cancer. 2006, 118, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta–catenin pathway and promotes EMT–like phenotype in trastuzumab–resistant HER2–overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Qiu, K.; Jia, Y. Modeling of mesenchymal hybrid epithelial state and phenotypic transitions in EMT and MET processes of cancer cells. Sci. Rep. 2018, 8, 14323. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben–Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben–Jacob, E. MicroRNA–based regulation of epithelial–hybrid–mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Jolly, M.K.; Tripathi, S.C.; Aguilar, M.; Hanash, S.M.; Levine, H.; Onuchic, J.N. Numb prevents a complete epithelial–mesenchymal transition by modulating Notch signalling. J. R. Soc. Interface 2017, 14, 20170512. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A mechanism–based computational model to capture the interconnections among epithelial–mesenchymal transition, cancer stem cells and Notch–Jagged signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef] [Green Version]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Que, J.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, B.; Wu, H.; Cai, J.; Sui, X.; Wang, Y.; Li, H.; Qiu, Y.; Wang, T.; Chen, Z.; et al. CD51 correlates with the TGF–beta pathway and is a functional marker for colorectal cancer stem cells. Oncogene 2017, 36, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Tsai, Y.P.; Wu, M.Z.; Teng, S.C.; Wu, K.J. Epigenetic reprogramming and post–transcriptional regulation during the epithelial–mesenchymal transition. Trends Genet. 2012, 28, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome–scale epigenetic reprogramming during epithelial–to–mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between HDAC3 and WDR5 is essential for hypoxia–induced epithelial–mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Ke, X.S.; Qu, Y.; Cheng, Y.; Li, W.C.; Rotter, V.; Oyan, A.M.; Kalland, K.H. Global profiling of histone and DNA methylation reveals epigenetic–based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genom. 2010, 11, 669. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial–mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.Y.; Tan, T.Z.; Ye, J.; Huang, R.L.; Lai, H.C.; Kappei, D.; Wollmann, H.; Guccione, E.; Huang, R.Y. The role of GRHL2 and epigenetic remodeling in epithelial–mesenchymal plasticity in ovarian cancer cells. Commun. Biol. 2019, 2, 272. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.F.; Liu, A.J.; Krishnakumar, R.; Freimer, J.W.; DeVeale, B.; Blelloch, R. GRHL2–dependent enhancer switching maintains a pluripotent stem cell transcriptional subnetwork after exit from naive pluripotency. Cell Stem Cell. 2018, 23, 226–238.e4. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.; Atkins, M.; Davie, K.; Imrichova, H.; Romanelli, L.; Christiaens, V.; Hulselmans, G.; Potier, D.; Wouters, J.; Taskiran, I.I.; et al. The transcription factor Grainy head primes epithelial enhancers for spatiotemporal activation by displacing nucleosomes. Nat. Genet. 2018, 50, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell–type–specific chromatin states differentially prime squamous cell carcinoma tumor–initiating cells for epithelial to mesenchymal transition. Cell Stem Cell. 2017, 20, 191–204.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Yang, W.H.; Lan, H.Y.; Huang, C.H.; Tai, S.K.; Tzeng, C.H.; Kao, S.Y.; Wu, K.J.; Hung, M.C.; Yang, M.H. RAC1 activation mediates Twist1–induced cancer cell migration. Nat. Cell Biol. 2012, 14, 366–374. [Google Scholar] [CrossRef]
- Liao, T.T.; Hsu, W.H.; Ho, C.H.; Hwang, W.L.; Lan, H.Y.; Lo, T.; Chang, C.C.; Tai, S.K.; Yang, M.H. let–7 modulates chromatin configuration and target gene repression through regulation of the arid3b complex. Cell Rep. 2016, 14, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno–Bueno, G.; Acloque, H.; Vega, S.; Barrallo–Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem–cell–like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben–Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [Green Version]
- Kroger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, T.-T.; Yang, M.-H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. https://doi.org/10.3390/cells9030623
Liao T-T, Yang M-H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells. 2020; 9(3):623. https://doi.org/10.3390/cells9030623
Chicago/Turabian StyleLiao, Tsai-Tsen, and Muh-Hwa Yang. 2020. "Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms" Cells 9, no. 3: 623. https://doi.org/10.3390/cells9030623
APA StyleLiao, T. -T., & Yang, M. -H. (2020). Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells, 9(3), 623. https://doi.org/10.3390/cells9030623