Scalable Generation of Mesenchymal Stem Cells and Adipocytes from Human Pluripotent Stem Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of Human Pluripotent Stem Cells
2.2. Differentiation of Human Pluripotent Stem Cells into Mesenchymal Stem Cells (MSCs)
2.3. Differentiation of MSCs into Adipocytes
2.4. Oil Red O Staining and Quantification
2.5. Differentiation of MSCs into Osteocytes and Chondrocytes
2.6. Immunocytochemistry
2.7. Flow Cytometry
2.8. Cell Proliferation Assays
2.9. Cell Viability Assay
2.10. RNA Sequencing and Data Analysis
2.11. Real Time PCR Analysis
2.12. Enrichment Analysis
2.13. Statistical analysis
3. Results
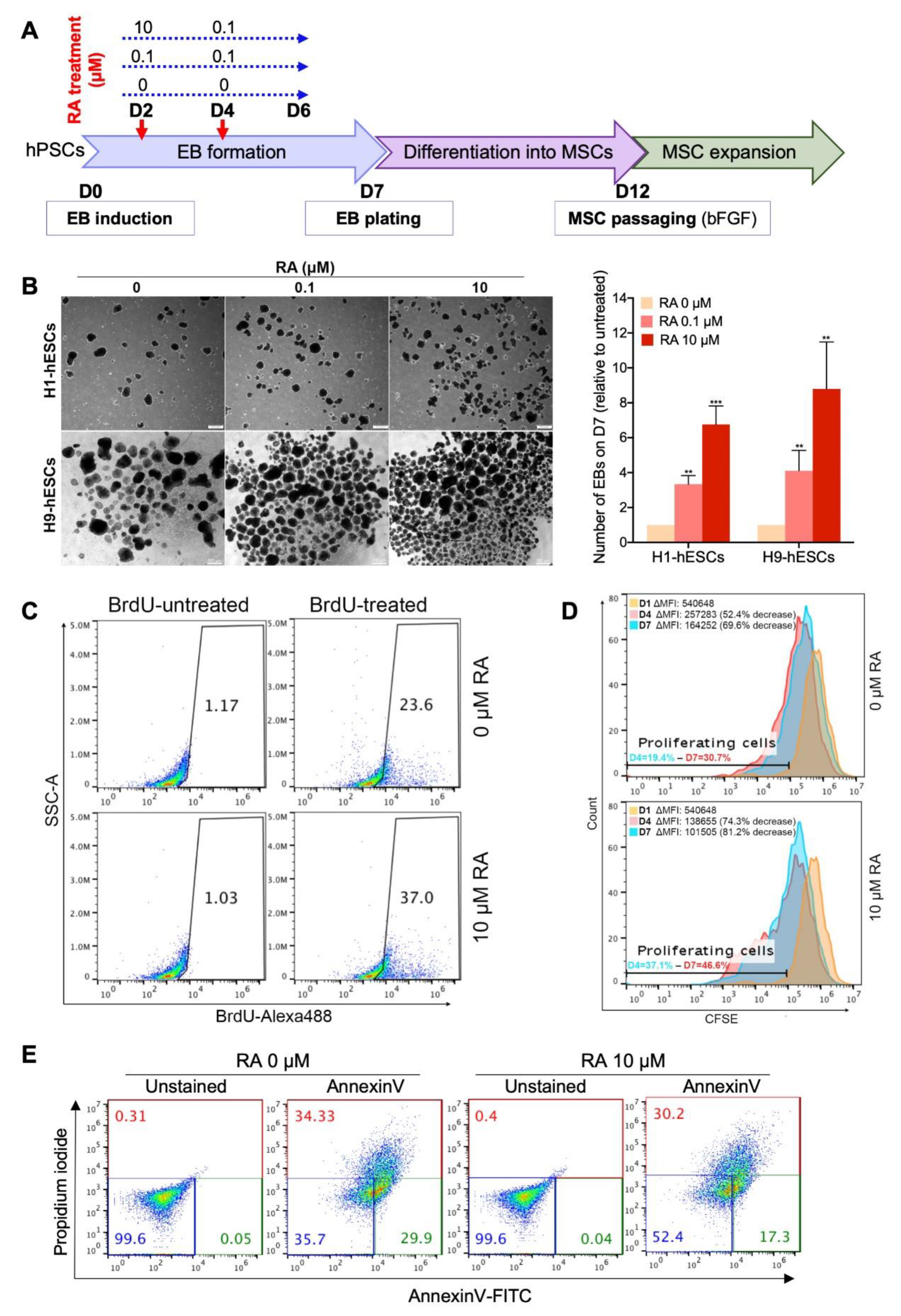
3.1. Short-Term RA Treatment Enhances Embryoid Body (EB) Formation from hPSCs by Inducing Both Cell Proliferation and Survival
3.2. RA-Mediated Inhibition of PSC-Derived EB Differentiation into MSCs Can Be Relieved by Cell Dissociation
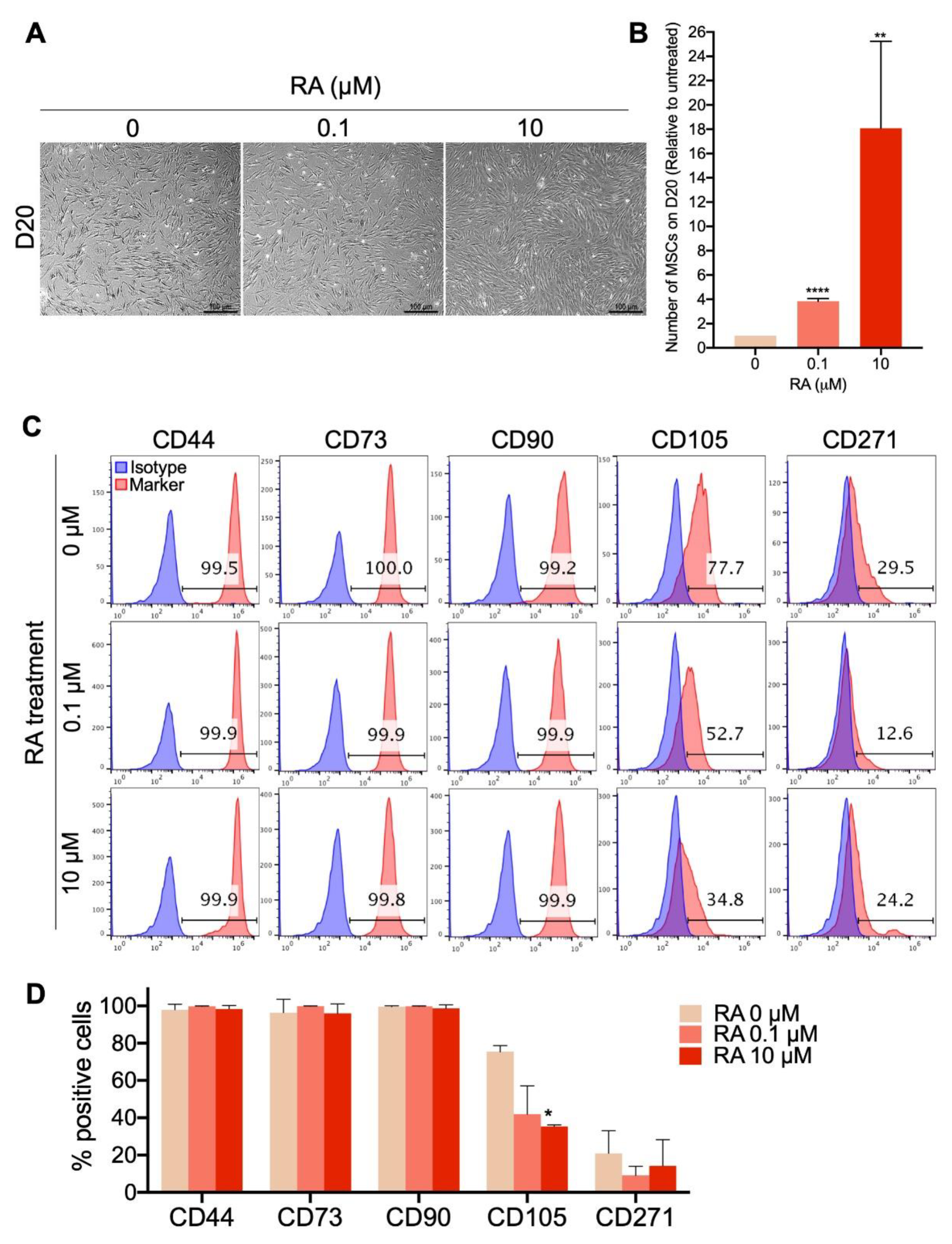
3.3. Short-Term EB Treatment with RA Strongly Enhances MSC Differentiation Yield
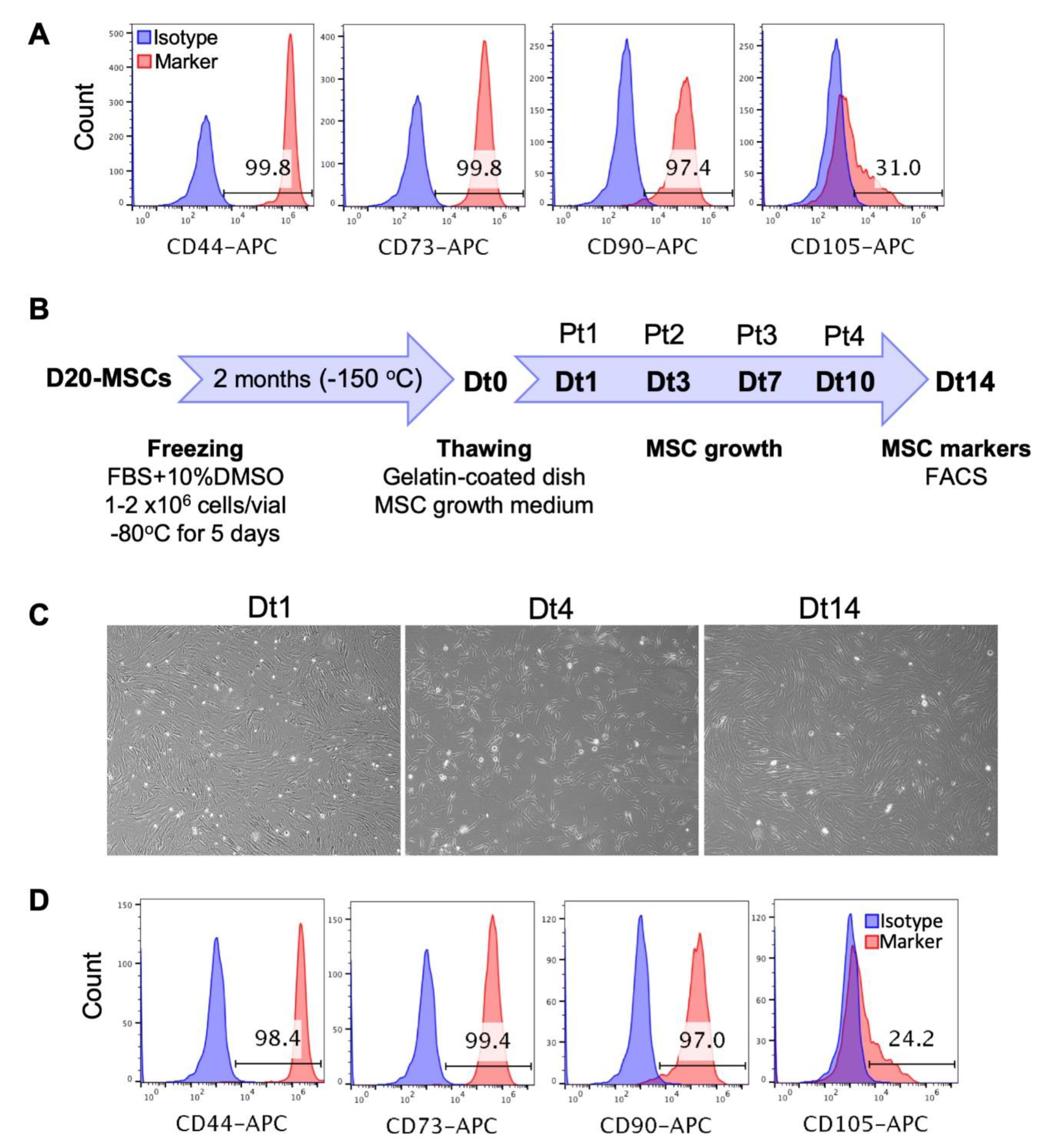
3.4. Long-Term Culture, Freezing and Thawing of the hPSC-Derived MSCs
3.5. Confirmation of the Multipotency of the hPSC-derived MSCs by Trilineage Differentiation
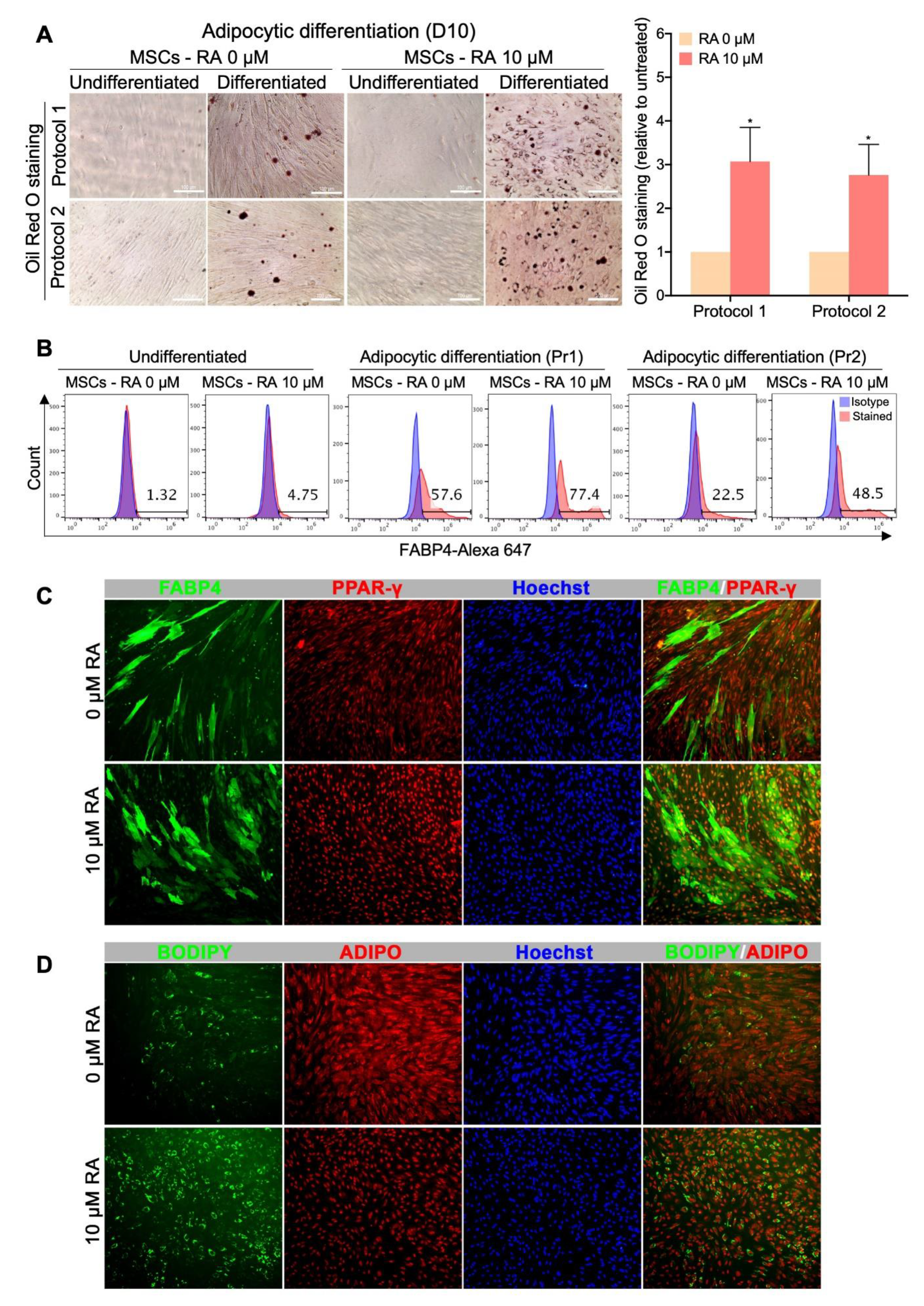
3.6. MSCs Derived from RA-treated EBs Display an Increased Adipocytic Differentiation Potential
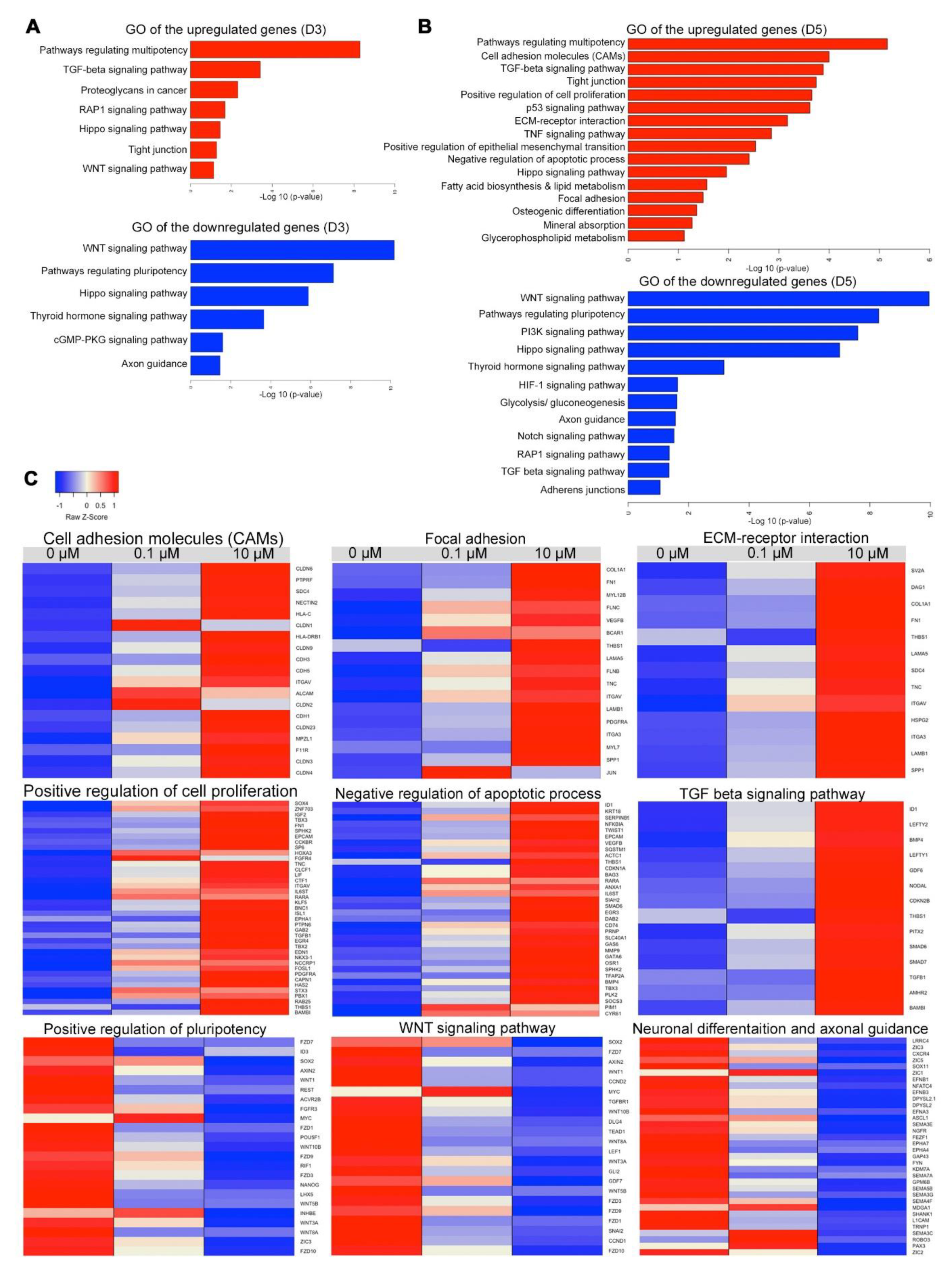
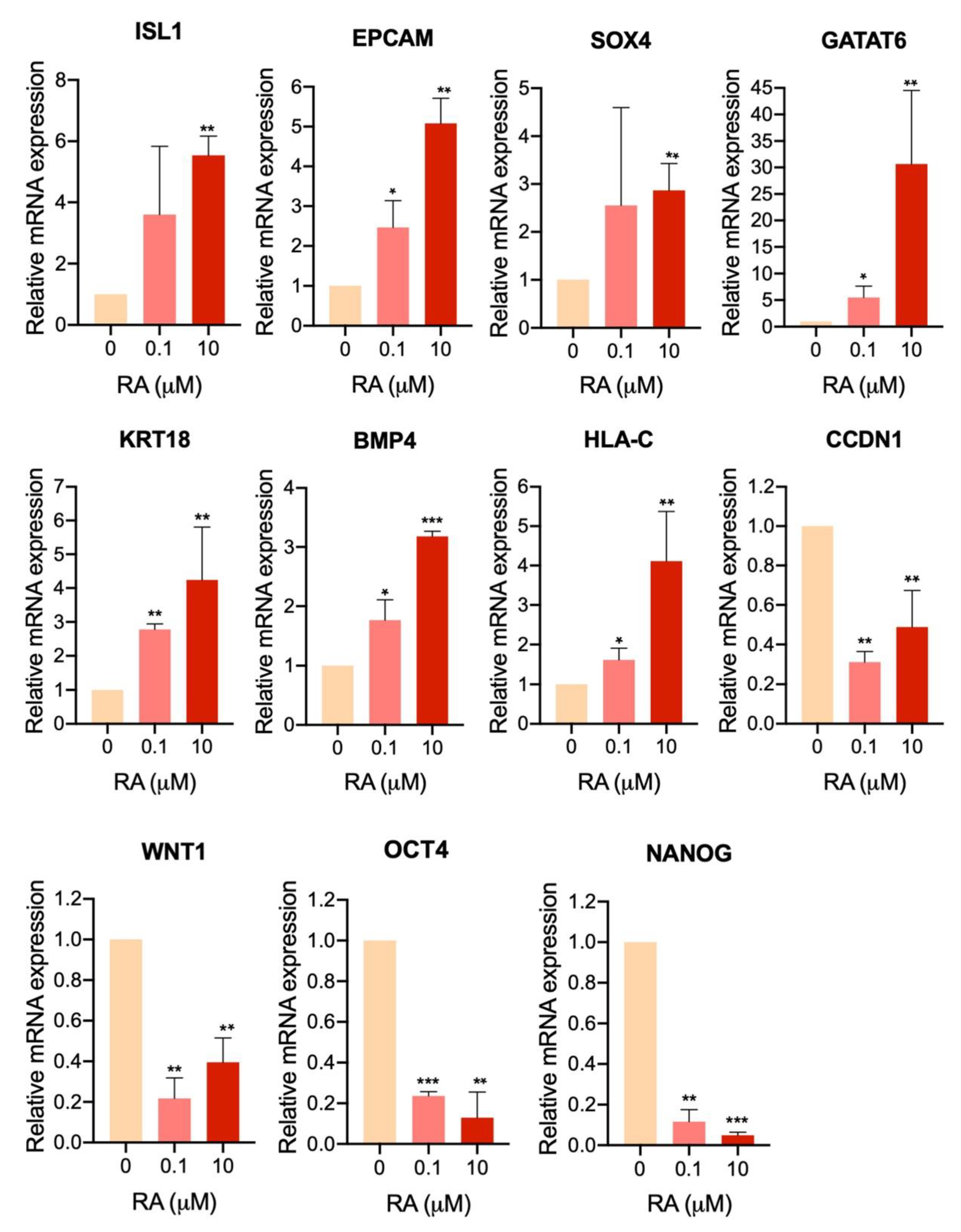
3.7. Transcriptome Profiling of Cells Treated with Different Concentrations of RA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEGs | differentially expressed genes |
| EBs | embryoid bodies |
| GO | Gene Ontology |
| hESCs | human embryonic stem cells |
| hiPSCs | human induced pluripotent stem cells |
| MSCs | mesenchymal stem cells |
| RA | All-trans-retinoic acid |
| RNA-seq | RNA-sequencing |
References
- Brown, C.; McKee, C.; Bakshi, S.; Walker, K.; Hakman, E.; Halassy, S.; Svinarich, D.; Dodds, R.; Govind, C.K.; Chaudhry, G.R. Mesenchymal stem cells: Cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 2019, 13, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE 2009, 4, e5846. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.T.; Squire, M.W.; Li, W.J. Characterization and evaluation of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Cell Tissue Res. 2014, 358, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Hematti, P. Derivation and immunological characterization of mesenchymal stromal cells from human embryonic stem cells. Exp. Hematol. 2008, 36, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Arpornmaeklong, P.; Brown, S.E.; Wang, Z.; Krebsbach, P.H. Phenotypic characterization, osteoblastic differentiation, and bone regeneration capacity of human embryonic stem cell-derived mesenchymal stem cells. Stem Cells Dev. 2009, 18, 955–968. [Google Scholar] [CrossRef]
- Barberi, T.; Willis, L.M.; Socci, N.D.; Studer, L. Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS Med. 2005, 2, e161. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- Van Harmelen, V.; Astrom, G.; Stromberg, A.; Sjolin, E.; Dicker, A.; Hovatta, O.; Ryden, M. Differential lipolytic regulation in human embryonic stem cell-derived adipocytes. Obesity (Silver Spring) 2007, 15, 846–852. [Google Scholar] [CrossRef]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, M.; Hosoda, K.; Nakane, M.; Mori, E.; Nakao, K.; Taura, D.; Yamamoto, Y.; Kusakabe, T.; Sone, M.; Sakurai, H.; et al. In vitro characterization and engraftment of adipocytes derived from human induced pluripotent stem cells and embryonic stem cells. Stem Cells Dev. 2013, 22, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Taura, D.; Noguchi, M.; Sone, M.; Hosoda, K.; Mori, E.; Okada, Y.; Takahashi, K.; Homma, K.; Oyamada, N.; Inuzuka, M.; et al. Adipogenic differentiation of human induced pluripotent stem cells: Comparison with that of human embryonic stem cells. FEBS Lett. 2009, 583, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clagett-Dame, M.; Knutson, D. Vitamin A in reproduction and development. Nutrients 2011, 3, 385–428. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar] [CrossRef]
- Mezquita, B.; Mezquita, C. Two Opposing Faces of Retinoic Acid: Induction of Stemness or Induction of Differentiation Depending on Cell-Type. Biomolecules 2019, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.T.; Parrotta, E.I.; Santamaria, G.; Cuda, G. Short-term retinoic acid treatment sustains pluripotency and suppresses differentiation of human induced pluripotent stem cells. Cell Death Dis. 2018, 9, 6. [Google Scholar] [CrossRef]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef]
- Dani, C.; Smith, A.G.; Dessolin, S.; Leroy, P.; Staccini, L.; Villageois, P.; Darimont, C.; Ailhaud, G. Differentiation of embryonic stem cells into adipocytes in vitro. J. Cell Sci. 1997, 110, 1279–1285. [Google Scholar]
- Ali, G.; Elsayed, A.K.; Nandakumar, M.; Bashir, M.; Younis, I.; Abu Aqel, Y.; Memon, B.; Temanni, R.; Abubaker, F.; Taheri, S.; et al. Keratinocytes Derived from Patient-Specific Induced Pluripotent Stem Cells Recapitulate the Genetic Signature of Psoriasis Disease. Stem Cells Dev. 2020. [Google Scholar] [CrossRef] [Green Version]
- Contador, D.; Ezquer, F.; Espinosa, M.; Arango-Rodriguez, M.; Puebla, C.; Sobrevia, L.; Conget, P. Dexamethasone and rosiglitazone are sufficient and necessary for producing functional adipocytes from mesenchymal stem cells. Exp. Biol. Med. 2015, 240, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, H.; Mir, L.M.; Andre, F.M. In vitro osteoblastic differentiation of mesenchymal stem cells generates cell layers with distinct properties. Stem Cell Res. Ther. 2018, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Memon, B.; Karam, M.; Al-Khawaga, S.; Abdelalim, E.M. Enhanced differentiation of human pluripotent stem cells into pancreatic progenitors co-expressing PDX1 and NKX6.1. Stem Cell Res. Ther. 2018, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedzhov, I.; Zernicka-Goetz, M. Self-organizing properties of mouse pluripotent cells initiate morphogenesis upon implantation. Cell 2014, 156, 1032–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Bennett, S.A.; Wang, L. Role of E-cadherin and other cell adhesion molecules in survival and differentiation of human pluripotent stem cells. Cell Adh. Migr. 2012, 6, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Narva, E.; Stubb, A.; Guzman, C.; Blomqvist, M.; Balboa, D.; Lerche, M.; Saari, M.; Otonkoski, T.; Ivaska, J. A Strong Contractile Actin Fence and Large Adhesions Direct Human Pluripotent Colony Morphology and Adhesion. Stem Cell Rep. 2017, 9, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Gregory, C.A.; Lee, R.H.; Reger, R.L.; Qin, L.; Hai, B.; Park, M.S.; Yoon, N.; Clough, B.; McNeill, E.; et al. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc. Natl. Acad. Sci. USA 2015, 112, 530–535. [Google Scholar] [CrossRef] [Green Version]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [Green Version]
- Ballim, R.D.; Mendelsohn, C.; Papaioannou, V.E.; Prince, S. The ulnar-mammary syndrome gene, Tbx3, is a direct target of the retinoic acid signaling pathway, which regulates its expression during mouse limb development. Mol. Biol. Cell 2012, 23, 2362–2372. [Google Scholar] [CrossRef]
- Boskovic, G.; Niles, R.M. T-box binding protein type two (TBX2) is an immediate early gene target in retinoic-acid-treated B16 murine melanoma cells. Exp. Cell Res. 2004, 295, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Higashihori, N.; Song, Y.; Richman, J.M. Expression and regulation of the decoy bone morphogenetic protein receptor BAMBI in the developing avian face. Dev. Dyn. 2008, 237, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Saavalainen, K.; Pasonen-Seppanen, S.; Dunlop, T.W.; Tammi, R.; Tammi, M.I.; Carlberg, C. The human hyaluronan synthase 2 gene is a primary retinoic acid and epidermal growth factor responding gene. J. Biol. Chem. 2005, 280, 14636–14644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villano, C.M.; Murphy, K.A.; Akintobi, A.; White, L.A. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in A2058 melanoma cells. Toxicol. Appl. Pharmacol. 2006, 210, 212–224. [Google Scholar] [CrossRef]
- Akiyama, H.; Tanaka, T.; Maeno, T.; Kanai, H.; Kimura, Y.; Kishi, S.; Kurabayashi, M. Induction of VEGF gene expression by retinoic acid through Sp1-binding sites in retinoblastoma Y79 cells. Invest. Ophthalmol. Vis. Sci. 2002, 43, 1367–1374. [Google Scholar]
- Kim, S.A.; Kang, J.H.; Cho, I.; Bae, S.W.; Hong, K.J. Cell-type specific regulation of thrombospondin-1 expression and its promoter activity by regulatory agents. Exp. Mol. Med. 2001, 33, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, S.; Meganathan, K.; Wagh, V.; Natarajan, K.; Hescheler, J.; Sachinidis, A. All-trans retinoic acid and basic fibroblast growth factor synergistically direct pluripotent human embryonic stem cells to extraembryonic lineages. Stem Cell Res. 2013, 10, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.S.; Bogart, D.F.; Castaneda, J.M.; Li, P.; Lupu, R. Cyr61 promotes breast tumorigenesis and cancer progression. Oncogene 2002, 21, 8178–8185. [Google Scholar] [CrossRef] [Green Version]
- Larsen, F.G.; Voorhees, J.J.; Astrom, A. Retinoic acid induces expression of early growth response gene-1 (Egr-1) in human skin in vivo and in cultured skin fibroblasts. J. Investig. Dermatol. 1994, 102, 730–733. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.C.; Jin, H.; Majumdar, A.; Noy, N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 2011, 108, 4340–4345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trocoli, A.; Bensadoun, P.; Richard, E.; Labrunie, G.; Merhi, F.; Schlafli, A.M.; Brigger, D.; Souquere, S.; Pierron, G.; Pasquet, J.M.; et al. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ. 2014, 21, 1852–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, L.; Bohnsack, B.L.; Niederreither, K.; Hirschi, K.K. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 2003, 130, 6465–6474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanchevalap, S.; Nandan, M.O.; Merlin, D.; Yang, V.W. All-trans retinoic acid inhibits proliferation of intestinal epithelial cells by inhibiting expression of the gene encoding Kruppel-like factor 5. FEBS Lett. 2004, 578, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, B.H.; Wang, S.; Moalim-Nour, L.; Mohib, K.; Lohnes, D.; Wang, L. Individual cell movement, asymmetric colony expansion, rho-associated kinase, and E-cadherin impact the clonogenicity of human embryonic stem cells. Biophys. J. 2010, 98, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, S.; Jezierski, A.; Moalim-Nour, L.; Mohib, K.; Parks, R.J.; Retta, S.F.; Wang, L. A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem Cells 2010, 28, 247–257. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, D.; Wei, B.; Jiang, J.; Kang, J.; Ling, K.; Gu, Y.; Li, J.; Xiao, L.; Pei, G. E-cadherin-mediated cell-cell contact is critical for induced pluripotent stem cell generation. Stem Cells 2010, 28, 1315–1325. [Google Scholar] [CrossRef]
- McGrath, M.; Tam, E.; Sladkova, M.; AlManaie, A.; Zimmer, M.; de Peppo, G.M. GMP-compatible and xeno-free cultivation of mesenchymal progenitors derived from human-induced pluripotent stem cells. Stem Cell Res. Ther. 2019, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Dong, L.; Wang, Y.; Zhang, X.; Yan, J. Lats1/2-Mediated Alteration of Hippo Signaling Pathway Regulates the Fate of Bone Marrow-Derived Mesenchymal Stem Cells. Biomed. Res. Int. 2018, 2018, 4387932. [Google Scholar] [CrossRef]
- Wang, Y.K.; Chen, C.S. Cell adhesion and mechanical stimulation in the regulation of mesenchymal stem cell differentiation. J. Cell Mol. Med. 2013, 17, 823–832. [Google Scholar] [CrossRef] [PubMed]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Christodoulides, C.; Lagathu, C.; Sethi, J.K.; Vidal-Puig, A. Adipogenesis and WNT signalling. Trends Endocrinol. Metab. 2009, 20, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaragosi, L.E.; Wdziekonski, B.; Fontaine, C.; Villageois, P.; Peraldi, P.; Dani, C. Effects of GSK3 inhibitors on in vitro expansion and differentiation of human adipose-derived stem cells into adipocytes. BMC Cell Biol. 2008, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlund, A.; Mejhert, N.; Lorente-Cebrian, S.; Astrom, G.; Dahlman, I.; Laurencikiene, J.; Ryden, M. Characterization of the Wnt inhibitors secreted frizzled-related proteins (SFRPs) in human adipose tissue. J. Clin. Endocrinol. Metab. 2013, 98, E503–E508. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Anderson, P.; Carrillo-Galvez, A.B.; Garcia-Perez, A.; Cobo, M.; Martin, F. CD105 (endoglin)-negative murine mesenchymal stromal cells define a new multipotent subpopulation with distinct differentiation and immunomodulatory capacities. PLoS ONE 2013, 8, e76979. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karam, M.; Younis, I.; Elareer, N.R.; Nasser, S.; Abdelalim, E.M. Scalable Generation of Mesenchymal Stem Cells and Adipocytes from Human Pluripotent Stem Cells. Cells 2020, 9, 710. https://doi.org/10.3390/cells9030710
Karam M, Younis I, Elareer NR, Nasser S, Abdelalim EM. Scalable Generation of Mesenchymal Stem Cells and Adipocytes from Human Pluripotent Stem Cells. Cells. 2020; 9(3):710. https://doi.org/10.3390/cells9030710
Chicago/Turabian StyleKaram, Manale, Ihab Younis, Noor R. Elareer, Sara Nasser, and Essam M. Abdelalim. 2020. "Scalable Generation of Mesenchymal Stem Cells and Adipocytes from Human Pluripotent Stem Cells" Cells 9, no. 3: 710. https://doi.org/10.3390/cells9030710
APA StyleKaram, M., Younis, I., Elareer, N. R., Nasser, S., & Abdelalim, E. M. (2020). Scalable Generation of Mesenchymal Stem Cells and Adipocytes from Human Pluripotent Stem Cells. Cells, 9(3), 710. https://doi.org/10.3390/cells9030710