Inhibition of Gastrin-Releasing Peptide Attenuates Phosphate-Induced Vascular Calcification

 ,
,
Abstract
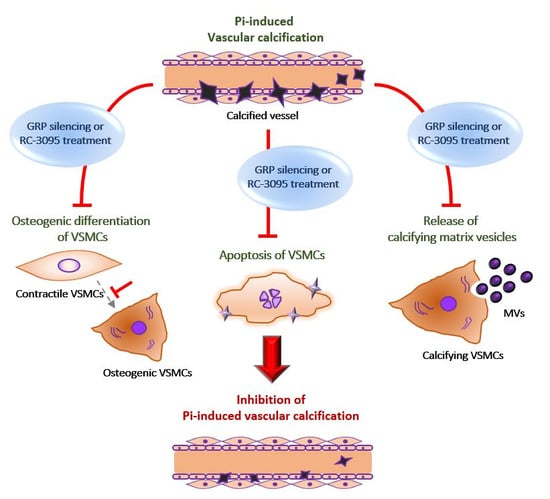
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Isolation and Culture
2.3. Calcification Induction and Quantification
2.4. Alizarin Red S Staining
2.5. Von Kossa Staining
2.6. Analysis of Calcification
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Quantitative Real-Time RT-PCR
2.9. Western Blot Analysis
2.10. Gene Knockdown by Small Interfering RNA
2.11. Immunocytochemistry
2.12. Flow Cytometry Analysis
2.13. TUNEL Assay
2.14. Matrix Vesicle Isolation
2.15. Arterial Ring Calcification
2.16. Induction of Chronic Kidney Disease (CKD) in the Mice Model
2.17. Statistical Analysis
3. Results
3.1. Elevated Expression of GRP and GRP Receptor in Pi-Induced VSMC Calcification
3.2. Inhibition of GRP Attenuates Pi–Induced Osteogenic Differentiation of VSMCs
3.3. Inhibition of GRP Ameliorates Pi–Induced Apoptosis and Matrix Vesicle Release of VSMCs
3.4. RC-3095 Suppresses Vascular Calcification in Ex Vivo Aortic Culture
3.5. RC-3095 Alleviates Aortic Calcification In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giachelli, C.M.; Speer, M.Y.; Li, X.; Rajachar, R.M.; Yang, H. Regulation of Vascular Calcification: Roles of Phosphate and Osteopontin. Circ. Res. 2005, 96, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular Calcification: Mechanisms and Clinical Ramifications. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linefsky, J.P.; O’Brien, K.D.; Katz, R.; de Boer, I.H.; Barasch, E.; Jenny, N.S.; Siscovick, D.S.; Kestenbaum, B. Association of Serum Phosphate Levels with Aortic Valve Sclerosis and Annular Calcification: The Cardiovascular Health Study. J. Am. Coll. Cardiol. 2011, 58, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachelli, C.M. The Emerging Role of Phosphate in Vascular Calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.S.; Cai, J.; Towler, D.A. Molecular Mechanisms of Vascular Calcification: Lessons Learned from the Aorta. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.D.; Chen, X.; Moe, S.M. Annexin-Mediated Matrix Vesicle Calcification in Vascular Smooth Muscle Cells. J. Bone Miner. Res. 2008, 23, 1798–1805. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis Accelerates Medial Vascular Calcification in Part by Triggering Smooth Muscle Cell Apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [Green Version]
- Patel, O.; Shulkes, A.; Baldwin, G.S. Gastrin-Releasing Peptide and Cancer. Biochim. Biophys. Acta 2006, 1766, 23–41. [Google Scholar] [CrossRef]
- Preston, S.R.; Miller, G.V.; Primrose, J.N. Bombesin-Like Peptides and Cancer. Crit. Rev. Oncol. Hematol. 1996, 23, 225–238. [Google Scholar] [CrossRef]
- Ohki-Hamazaki, H.; Iwabuchi, M.; Maekawa, F. Development and Function of Bombesin-Like Peptides and their Receptors. Int. J. Dev. Biol. 2005, 49, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; Park, H.J.; Kim, Y.; Kim, H.J.; Bae, S.K.; Bae, M.K. Gastrin-Releasing Peptide Induces Monocyte Adhesion to Vascular Endothelium by Upregulating Endothelial Adhesion Molecules. Biochem. Biophys. Res. Commun. 2017, 485, 542–549. [Google Scholar] [PubMed]
- Park, H.J.; Kim, M.K.; Kim, Y.; Bae, S.S.; Kim, H.J.; Bae, S.K.; Bae, M.K. Gastrin-Releasing Peptide Promotes the Migration of Vascular Smooth Muscle Cells through Upregulation of Matrix Metalloproteinase-2 and -9. BMB Rep. 2017, 50, 628–633. [Google Scholar] [PubMed] [Green Version]
- Petronilho, F.; Danielski, L.G.; Roesler, R.; Schwartsmann, G.; Dal-Pizzol, F. Gastrin-Releasing Peptide as a Molecular Target for Inflammatory Diseases: An Update. Inflamm. Allergy Drug Targets 2013, 12, 172–177. [Google Scholar] [CrossRef]
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory Mechanisms in Vascular Calcification. Nat. Rev. Cardiol. 2010, 7, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Hohla, F.; Schally, A.V. Targeting Gastrin Releasing Peptide Receptors: New Options for the Therapy and Diagnosis of Cancer. Cell. Cycle 2010, 9, 1738–1741. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.V.; Steckert, A.V.; Mina, F.; Petronilho, F.; Roesler, R.; Schwartsmann, G.; Ritter, C.; Dal-Pizzol, F. Effects of an Antagonist of the Gastrin-Releasing Peptide Receptor in an Animal Model of Uveitis. Invest. Ophthalmol. Vis. Sci. 2009, 50, 5300–5303. [Google Scholar] [CrossRef] [Green Version]
- Cornelio, D.B.; Dal-Pizzol, F.; Roesler, R.; Schwartsmann, G. Targeting the bombesin/gastrin-Releasing Peptide Receptor to Treat Sepsis. Recent. Pat. Antiinfect Drug Discov. 2007, 2, 178–181. [Google Scholar] [CrossRef]
- Oliveira, P.G.; Grespan, R.; Pinto, L.G.; Meurer, L.; Brenol, J.C.; Roesler, R.; Schwartsmann, G.; Cunha, F.Q.; Xavier, R.M. Protective Effect of RC-3095, an Antagonist of the Gastrin-Releasing Peptide Receptor, in Experimental Arthritis. Arthritis Rheum. 2011, 63, 2956–2965. [Google Scholar] [CrossRef]
- Jonkman, J.E.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An Introduction to the Wound Healing Assay using Live-Cell Microscopy. Cell. Adh Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Kim, C.; Chae, S.W.; Oh, S. Unsupervised Segmentation of Overlapped Nuclei using Bayesian Classification. IEEE Trans. Biomed. Eng. 2010, 57, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, T.; Orimo, H.; Shimizu, A.; Tsuruoka, S. Development of a Novel Chronic Kidney Disease Mouse Model to Evaluate the Progression of Hyperphosphatemia and Associated Mineral Bone Disease. Sci. Rep. 2017, 7, 2233. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate Regulation of Vascular Smooth Muscle Cell Calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Miyazono, K. Signal Transduction by Bone Morphogenetic Protein Receptors: Functional Roles of Smad Proteins. Bone 1999, 25, 91–93. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y.P. TGF-Beta and BMP Signaling in Osteoblast, Skeletal Development, and Bone Formation, Homeostasis and Disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Clarke, M.C.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic Apoptosis of Vascular Smooth Muscle Cells Accelerates Atherosclerosis and Promotes Calcification and Medial Degeneration. Circ. Res. 2008, 102, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Stehbens, W.E. The Significance of Programmed Cell Death Or Apoptosis and Matrix Vesicles in Atherogenesis. Cell. Mol. Biol. 2000, 46, 99–110. [Google Scholar]
- Golub, E.E. Role of Matrix Vesicles in Biomineralization. Biochim. Biophys. Acta 2009, 1790, 1592–1598. [Google Scholar]
- Akiyoshi, T.; Ota, H.; Iijima, K.; Son, B.K.; Kahyo, T.; Setou, M.; Ogawa, S.; Ouchi, Y.; Akishita, M. A Novel Organ Culture Model of Aorta for Vascular Calcification. Atherosclerosis 2016, 244, 51–58. [Google Scholar]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular Calcification in Animal Models of CKD: A Review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [PubMed]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial-Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprague, A.H.; Khalil, R.A. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [PubMed] [Green Version]
- Orr, A.W.; Hastings, N.E.; Blackman, B.R.; Wamhoff, B.R. Complex Regulation and Function of the Inflammatory Smooth Muscle Cell Phenotype in Atherosclerosis. J. Vasc. Res. 2010, 47, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [PubMed]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; Echeverri, D.; Truszczynska, H.; Sharma, S.K.; Badimon, J.J.; O’Connor, W.N. Plaque Neovascularization is Increased in Ruptured Atherosclerotic Lesions of Human Aorta: Implications for Plaque Vulnerability. Circulation 2004, 110, 2032–2038. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.; Zudaire, E.; Julian, M.; Moody, T.W.; Cuttitta, F. Gastrin-Releasing Peptide (GRP) Induces Angiogenesis and the Specific GRP Blocker 77427 Inhibits Tumor Growth in Vitro and in Vivo. Oncogene 2005, 24, 4106–4113. [Google Scholar]
- Leopold, J.A. Vascular Calcification: Mechanisms of Vascular Smooth Muscle Cell Calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Farzaneh-Far, A.; Shanahan, C.M.; Weissberg, P.L. The Role of Apoptosis in the Initiation of Vascular Calcification. Z. Kardiol. 2001, 90, 43–46. [Google Scholar]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification in Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zazzeroni, L.; Faggioli, G.; Pasquinelli, G. Mechanisms of Arterial Calcification: The Role of Matrix Vesicles. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, R.; Qin, X.; Wang, L.; Xiao, J.; Song, Y.; Sheng, X.; Guo, M.; Ji, X. Sp1 Plays an Important Role in Vascular Calcification both in Vivo and in Vitro. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X. Pathophysiology of Vascular Calcification in Chronic Kidney Disease. Circ. Res. 2004, 95, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Palit, S.; Kendrick, J. Vascular Calcification in Chronic Kidney Disease: Role of Disordered Mineral Metabolism. Curr. Pharm. Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef] [Green Version]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate Feeding Induces Arterial Medial Calcification in Uremic Mice: Role of Serum Phosphorus, Fibroblast Growth Factor-23, and Osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar]
- Rahman, A.; Yamazaki, D.; Sufiun, A.; Kitada, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. A Novel Approach to Adenine-Induced Chronic Kidney Disease Associated Anemia in Rodents. PLoS ONE 2018, 13, e0192531. [Google Scholar] [CrossRef]
- Yang, H.C.; Zuo, Y.; Fogo, A.B. Models of Chronic Kidney Disease. Drug Discov. Today Dis. Models 2010, 7, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Kukida, M.; Mogi, M.; Kan-No, H.; Tsukuda, K.; Bai, H.Y.; Shan, B.S.; Yamauchi, T.; Higaki, A.; Min, L.J.; Iwanami, J.; et al. AT2 Receptor Stimulation Inhibits Phosphate-Induced Vascular Calcification. Kidney Int. 2019, 95, 138–148. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Shao, J.S.; Lai, C.F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.L.; Towler, D.A. Aortic Msx2-Wnt Calcification Cascade is Regulated by TNF-Alpha-Dependent Signals in Diabetic Ldlr-/- Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.S.; Cheng, S.L.; Sadhu, J.; Towler, D.A. Inflammation and the Osteogenic Regulation of Vascular Calcification: A Review and Perspective. Hypertension 2010, 55, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Jahnen-Dechent, W.; Heiss, A.; Schafer, C.; Ketteler, M. Fetuin-A Regulation of Calcified Matrix Metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [PubMed] [Green Version]
- Parhami, F.; Morrow, A.D.; Balucan, J.; Leitinger, N.; Watson, A.D.; Tintut, Y.; Berliner, J.A.; Demer, L.L. Lipid Oxidation Products have Opposite Effects on Calcifying Vascular Cell and Bone Cell Differentiation. A Possible Explanation for the Paradox of Arterial Calcification in Osteoporotic Patients. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 680–687. [Google Scholar] [PubMed]
- Price, P.A.; June, H.H.; Buckley, J.R.; Williamson, M.K. Osteoprotegerin Inhibits Artery Calcification Induced by Warfarin and by Vitamin D. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1610–1616. [Google Scholar]
- Cornelio, D.B.; Roesler, R.; Schwartsmann, G. Gastrin-Releasing Peptide Receptor as a Molecular Target in Experimental Anticancer Therapy. Ann. Oncol. 2007, 18, 1457–1466. [Google Scholar] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-J.; Kim, Y.; Kim, M.-K.; Hwang, J.J.; Kim, H.J.; Bae, S.-K.; Bae, M.-K. Inhibition of Gastrin-Releasing Peptide Attenuates Phosphate-Induced Vascular Calcification. Cells 2020, 9, 737. https://doi.org/10.3390/cells9030737
Park H-J, Kim Y, Kim M-K, Hwang JJ, Kim HJ, Bae S-K, Bae M-K. Inhibition of Gastrin-Releasing Peptide Attenuates Phosphate-Induced Vascular Calcification. Cells. 2020; 9(3):737. https://doi.org/10.3390/cells9030737
Chicago/Turabian StylePark, Hyun-Joo, Yeon Kim, Mi-Kyoung Kim, Jae Joon Hwang, Hyung Joon Kim, Soo-Kyung Bae, and Moon-Kyoung Bae. 2020. "Inhibition of Gastrin-Releasing Peptide Attenuates Phosphate-Induced Vascular Calcification" Cells 9, no. 3: 737. https://doi.org/10.3390/cells9030737
APA StylePark, H. -J., Kim, Y., Kim, M. -K., Hwang, J. J., Kim, H. J., Bae, S. -K., & Bae, M. -K. (2020). Inhibition of Gastrin-Releasing Peptide Attenuates Phosphate-Induced Vascular Calcification. Cells, 9(3), 737. https://doi.org/10.3390/cells9030737