Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
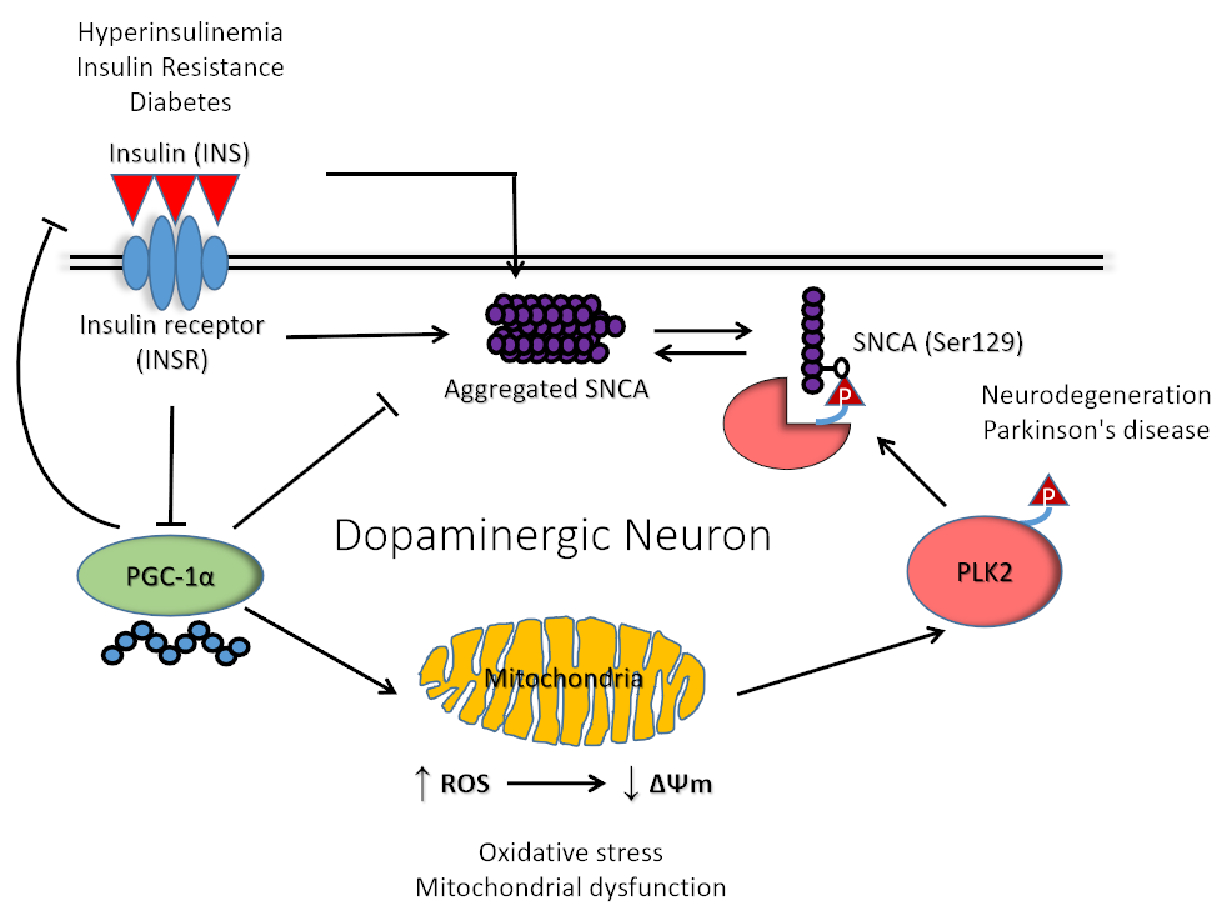
1. Introduction
2. Materials and Methods
2.1. Animal
2.2. Assessment of Motor Balance and Coordination, In Vivo
2.3. Adipocyte-Derived Stem Cell Isolation, Culture, and Neuronal Differentiation
2.4. Cell Culture
2.5. Fluorescence-Activated Cell Sorting Flow Cytometry
2.6. Western Blot Analysis
2.7. Protease K Digestion of α-Synuclein
2.8. Oxidative Stress Assessment
2.9. Mitochondrial Membrane Potential (ΔΨm) Assessment
2.10. Immunohistochemistry
2.11. Immunocytochemistry (IHC)
2.12. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time Polymerase Chain Reaction
2.13. Polo-Like Kinase 2 Activity Assays
2.14. Confocal Imaging
2.15. Deconvolution Microscopy Imaging
2.16. Datasets and Bioinformatics Analyses
2.17. Statistical Analysis
3. Results
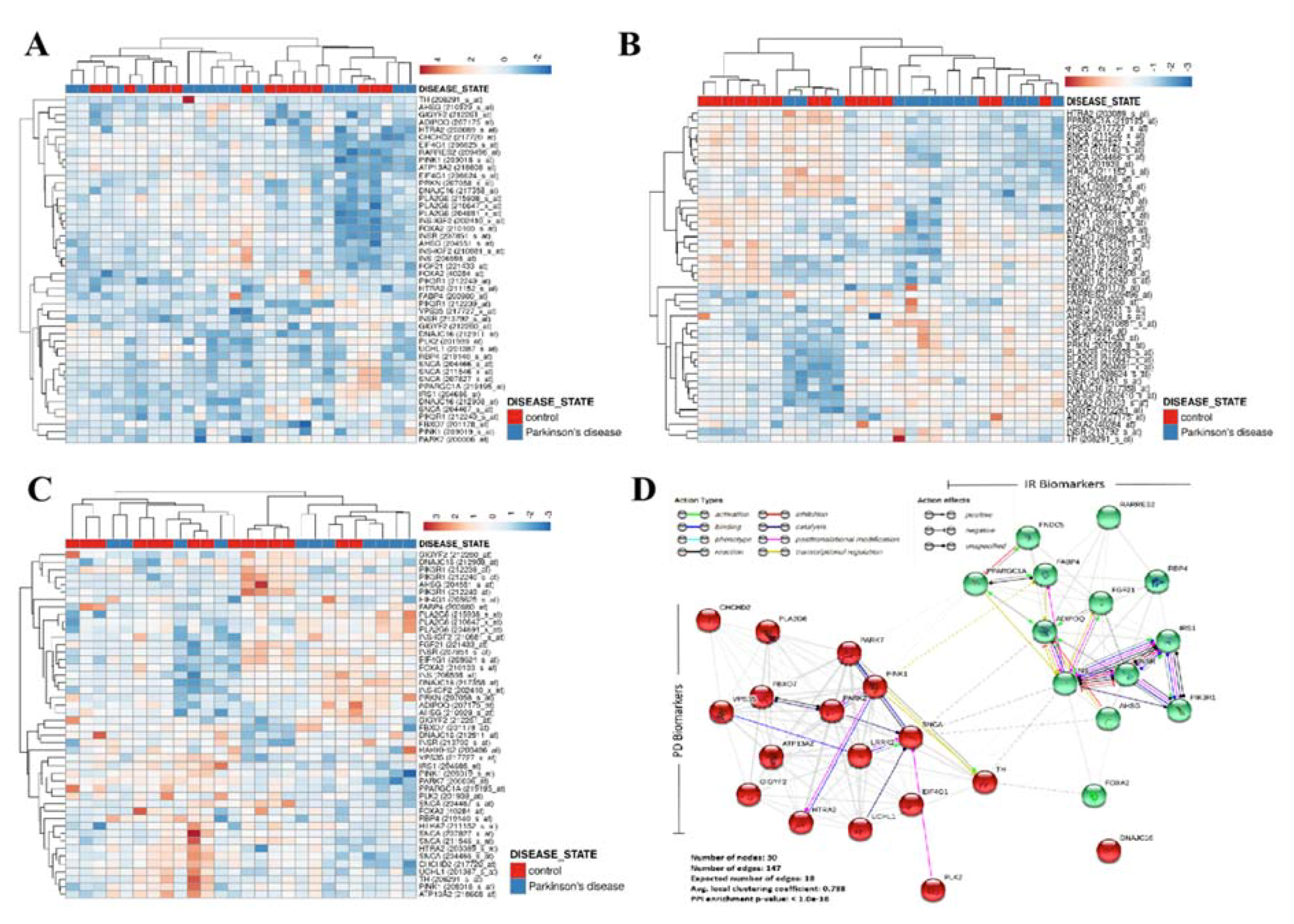
3.1. Patients’ Genomic Landscape Indicates a Nosological Association between Diabetes and PD, In Silico
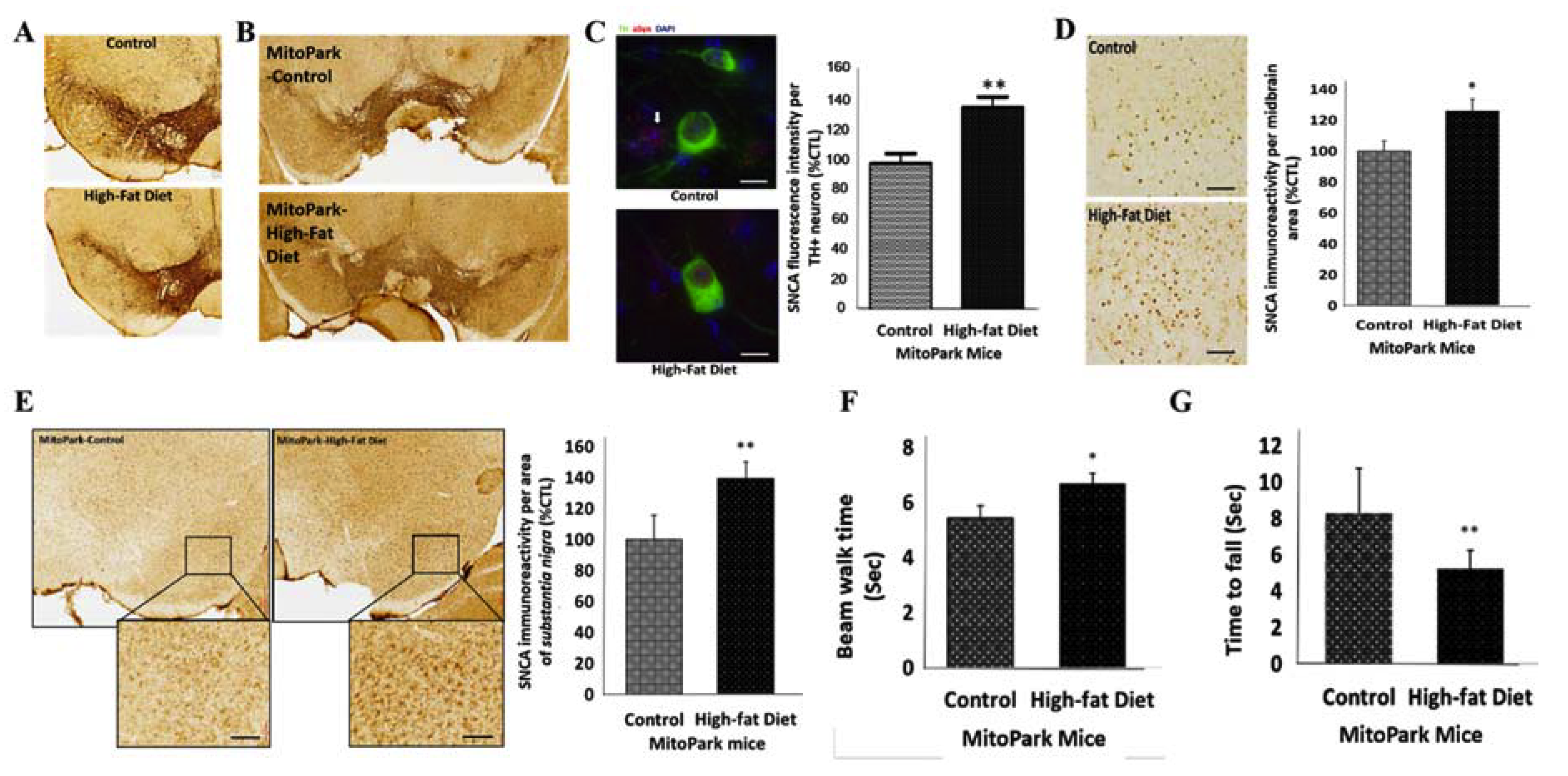
3.2. Diabetes Contributes to the Progression of PD by Amplifying the Loss of TH+ Dopaminergic Neurons and Enhancing SNCA Expression, In Vivo
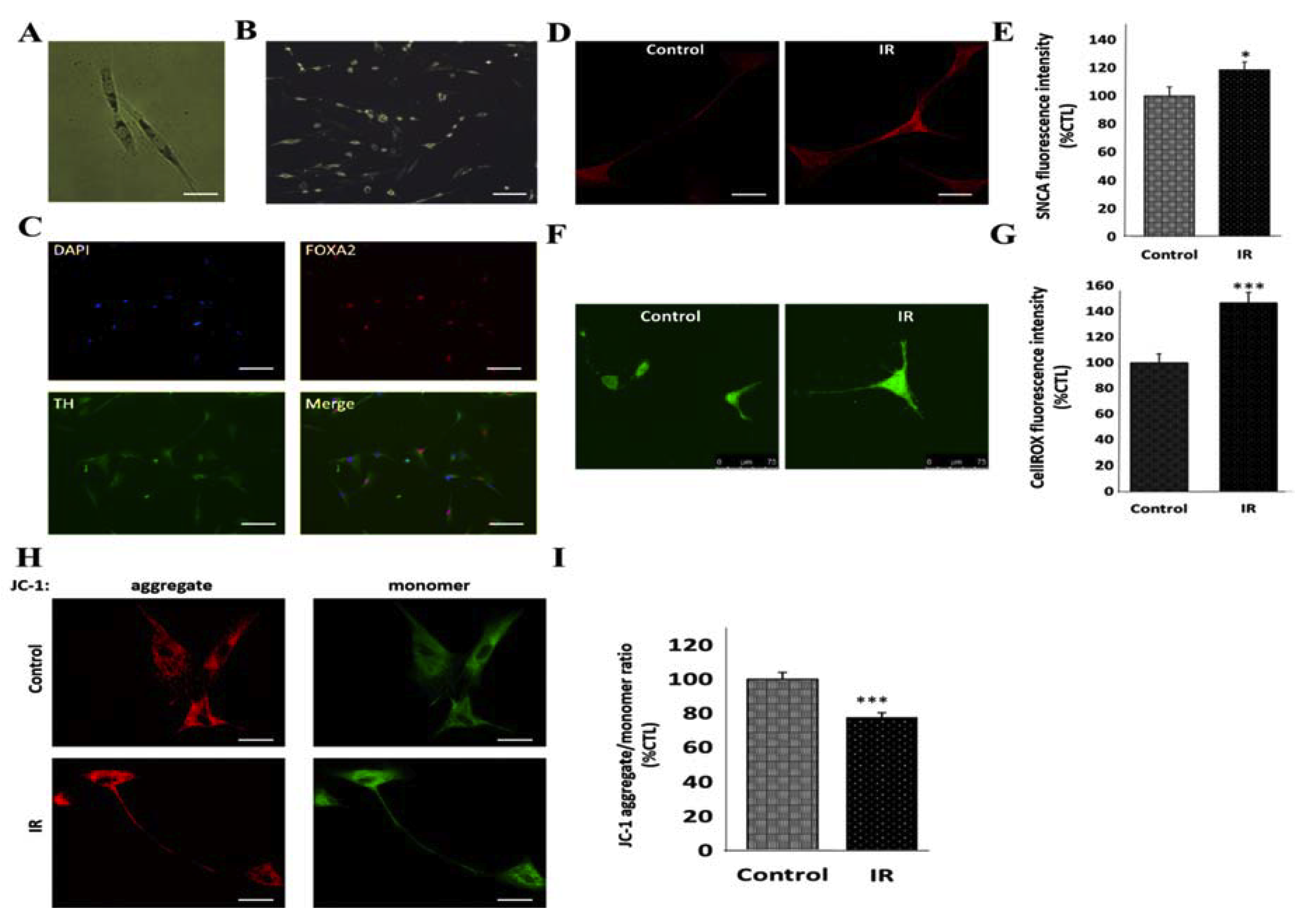
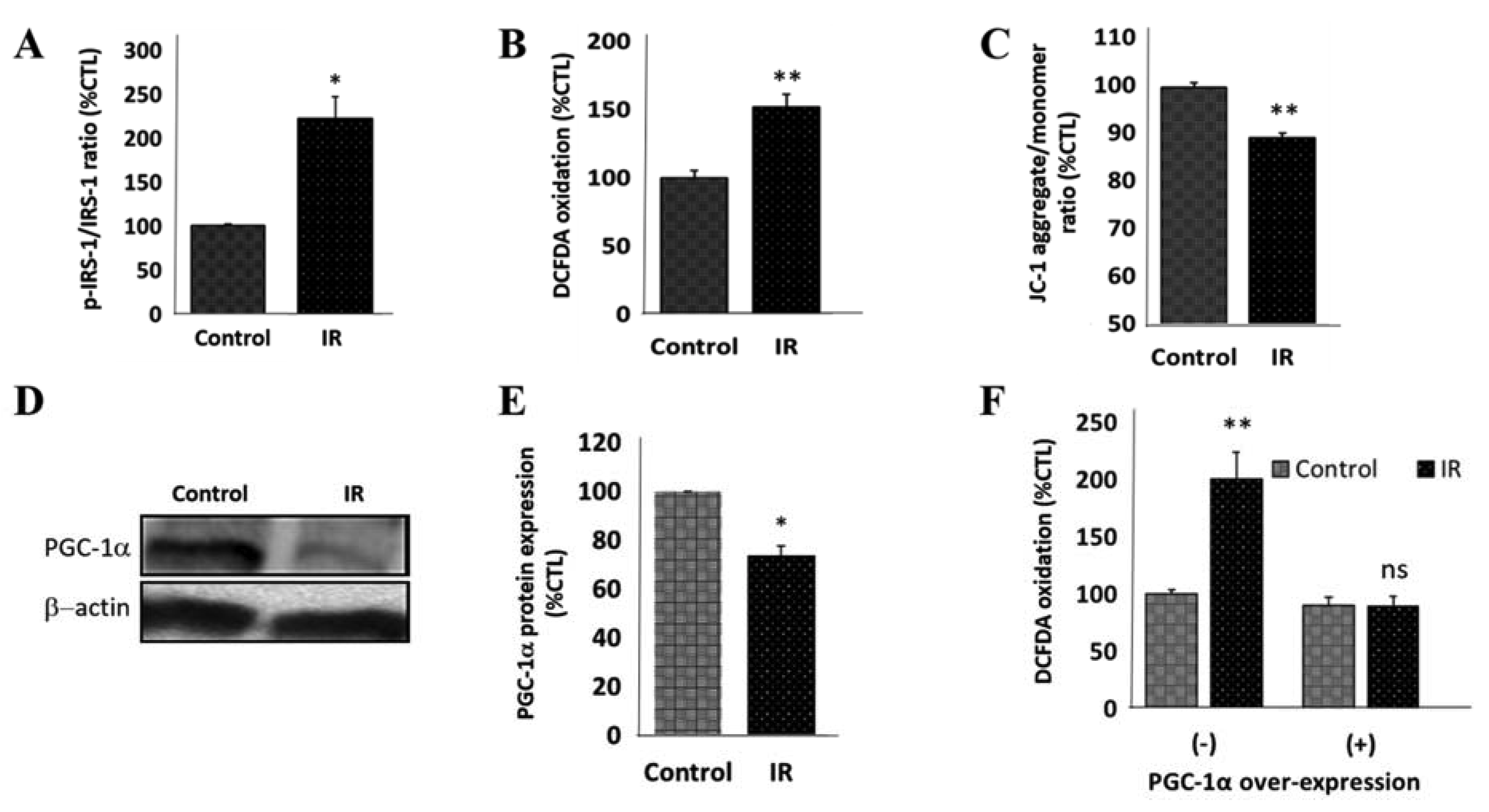
3.3. Insulin Resistance Is Associated with Increased Expression of SNCA and Mitochondrial Dysfunction in Human Differentiated Dopaminergic Neurons
3.4. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha (PPARGC1A/PGC-1α) Impedes IR-Induced Mitochondrial Dysfunction
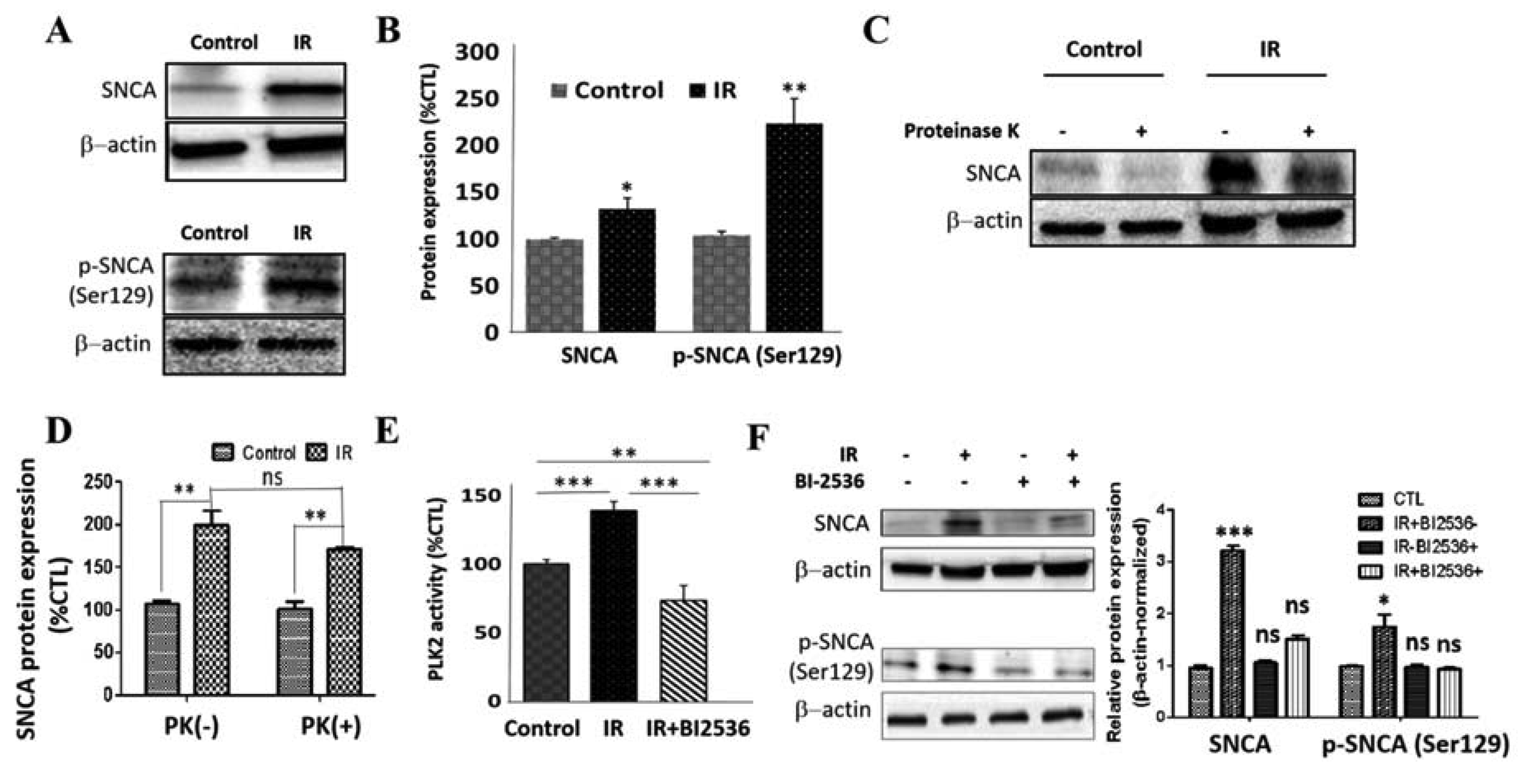
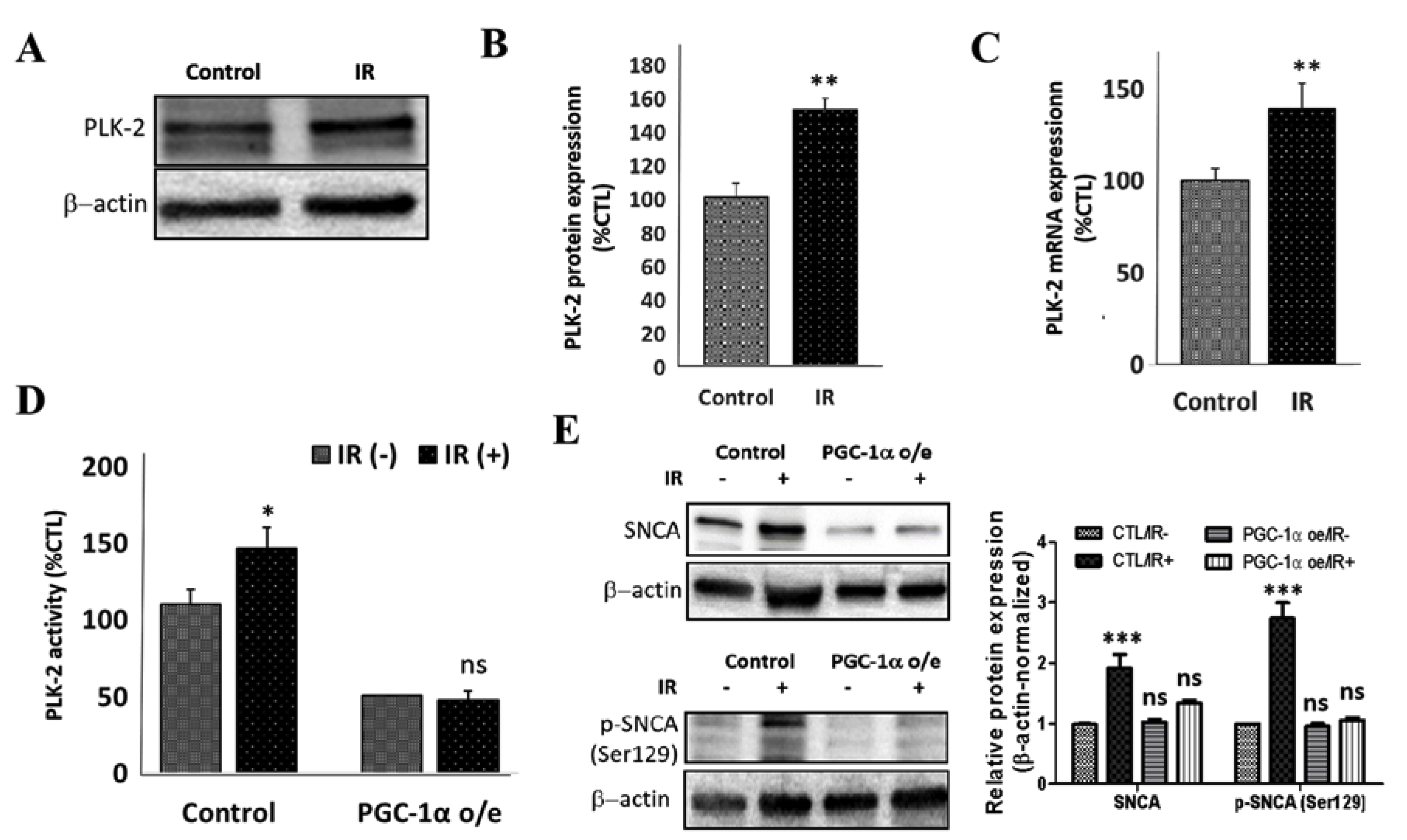
3.5. IR Elicits Increased Total, Ser129-Phosphorylated, and Proteinase K-Resistant SNCA through a Polo-Like Kinase 2-Dependent Mechanism
3.6. PGC-1α Overexpression Represses IR-Enhanced PLK2 Activity and Subsequent Increase in SNCA
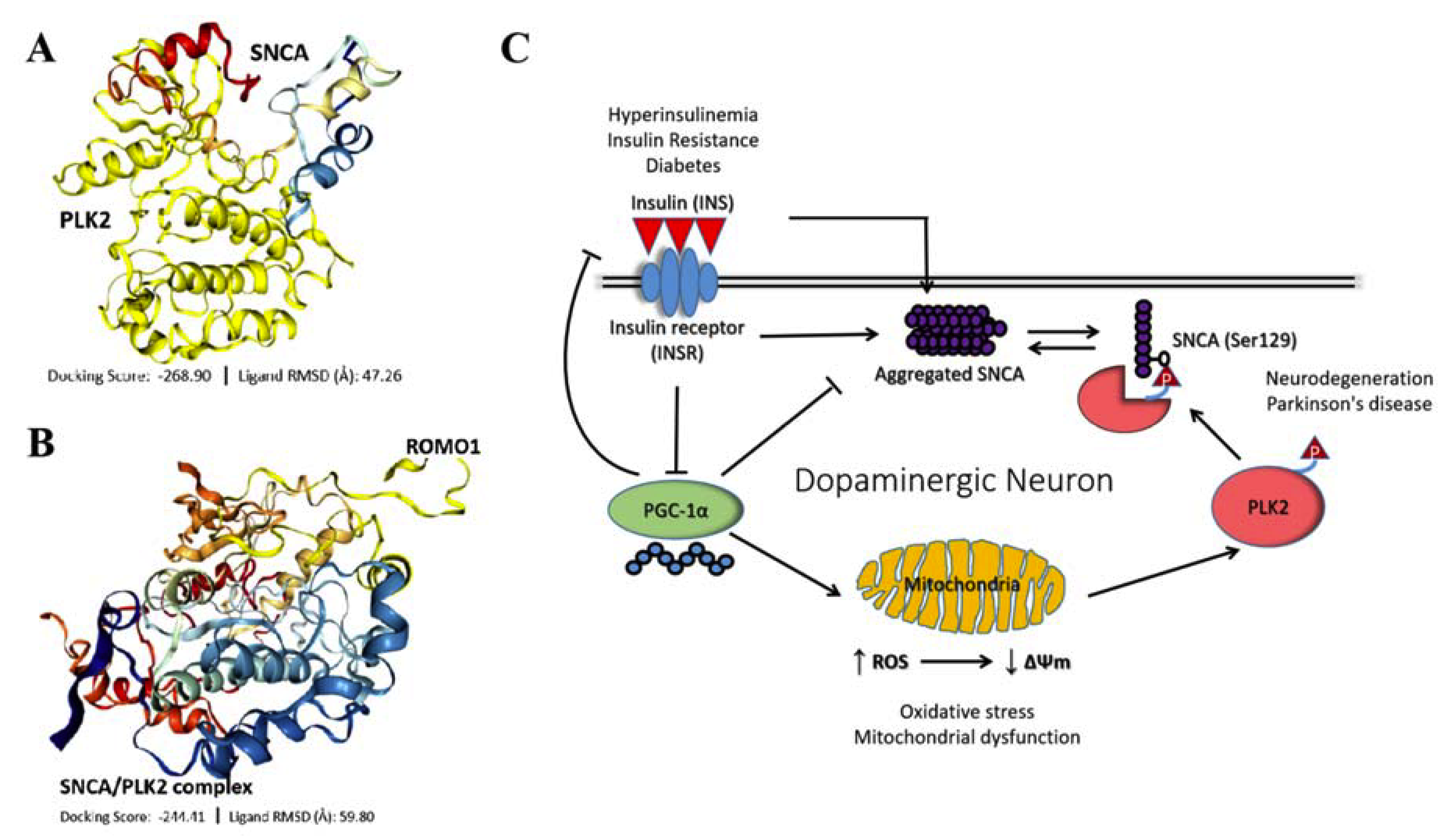
3.7. IR-Driven SNCA-PLK2-ROS Signaling Is Implicated in PD Development and Progression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signaling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Morsi, M.; Maher, A.; Aboelmagd, O.; Johar, D.; Bernstein, L. A shared comparison of diabetes mellitus and neurodegenerative disorders. J. Cell. Biochem. 2018, 119, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, H. Alzheimer’s disease could be “type 3 diabetes”. Lancet Neurol. 2006, 5, 388–389. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.; Hansen, J.; Rugbjerg, K.; Wermuth, L.; Ritz, B. Diabetes and the Risk of Developing Parkinson’s Disease in Denmark. Diabetes Care 2011, 34, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chang, Y.-H.; Chen, H.-F.; Su, Y.-H.; Su, H.-F.; Li, C.-Y. Risk of Parkinson Disease Onset in Patients with Diabetes. A 9-year population-based cohort study with age and sex stratifications. Diabetes Care 2012, 35, 1047–1049. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 Diabetes and the Risk of Parkinson’s Disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Fu, D.L.; Li, H.Q.; Liu, A.J.; Li, J.H.; Zheng, G.Q. Diabetes and risk of Parkinson’s disease: An updated meta-analysis of case-control studies. PLoS ONE 2014, 9, e85781. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Goldacre, R.; Pakpoor, J.; Noyce, A.J.; Warner, T.T. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018, 91, e139–e142. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, W.; Liu, F.T.; Li, J.Q.; Cao, X.P.; Tan, L.; Wang, J.; Yu, J. Modifiable risk factors for cognitive impairment in Parkinson’s disease: A systematic review and meta-analysis of prospective cohort studies. Mov. Disord. 2019, 34, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Zimmermann, J.; Sixel-Doring, F.; Focke, N.K.; Wicke, T.; Ebentheuer, J.; Schaumburg, M.; Lang, E.; Friede, T.; Trenkwalder, C. Baseline predictors for progression 4 years after Parkinson’s disease diagnosis in the De Novo Parkinson Cohort (DeNoPa). Mov. Disord. 2019, 34, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Crea, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Solmaz, V.; Köse Özlece, H.; Eroglu, H.A.; Aktuğ, H.; Erbaş, O.; Taşkıran, D. Accumulation of α-Synuclein in Cerebellar Purkinje Cells of Diabetic Rats and Its Potential Relationship with Inflammation and Oxidative Stress Markers. Neurol. Res. Int. 2017, 2017, 5952149. [Google Scholar] [CrossRef] [Green Version]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [Green Version]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Vrach, N.B.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse—An animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat. Disord. 2009, 15, S185. [Google Scholar] [CrossRef]
- Heydemann, A. An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 2902351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailey, B.; Hosseini, A.; Baker, J.; Young, A.; Alfonso, Z.; Hicok, K.; Wallace, A.M.; Cohen, S.R. Adipose-derived stem cells: Methods for isolation and applications for clinical use. Methods Mol. Biol. 2014, 1210, 161–181. [Google Scholar] [PubMed]
- Ivankovic, D.; Chau, K.-Y.; Schapira, A.H.V.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.A.; Labadorf, A.; Kennedy, N.J.; Han, M.S.; Yap, Y.S.; Matthews, B.; Xin, X.; Sun, L.; Davis, R.J.; Lodish, H.F.; et al. Analysis of in vitro insulin-resistance models and their physiological relevance to in vivo diet-induced adipose insulin resistance. Cell Rep. 2013, 5, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Reitboeck, P.; Anichtchik, O.; Dalley, J.W.; Ninkina, N.; Tofaris, G.K.; Buchman, V.L.; Spillantini, M.G. Endogenous alpha-synuclein influences the number of dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 2013, 248, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.T.; Chau, K.-Y.; Schapira, A.H.V. The Cytomegalovirus protein pUL37 × 1 targets mitochondria to mediate neuroprotection. Sci. Rep. 2016, 6, 31373. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.C. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat. Rec. 2013, 296, 378–381. [Google Scholar] [CrossRef]
- Stoessl, A.J.; Lehericy, S.; Strafella, A.P. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet 2014, 384, 532–544. [Google Scholar] [CrossRef] [Green Version]
- Park, S.E.; Park, C.Y.; Sweeney, G. Biomarkers of insulin sensitivity and insulin resistance: Past, present and future. Crit. Rev. Clin. Lab. Sci. 2015, 52, 180–190. [Google Scholar] [CrossRef]
- Morris, J.K.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Neurodegeneration in an animal model of Parkinson’s disease is exacerbated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1082–R1090. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Y.; Beavan, M.; Chau, K.Y.; Taanman, J.W.; Schapira, A.H.V. A Human Neural Crest Stem Cell-Derived Dopaminergic Neuronal Model Recapitulates Biochemical Abnormalities in GBA1 Mutation Carriers. Stem Cell Rep. 2017, 8, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajan, M.; Hansen, B.; Ivey, R., III; Sajan, J.; Ari, C.; Song, S.; Braun, U.; Leitges, M.; Farese-Higgs, M.; Farese, R.V. Brain Insulin Signaling Is Increased in Insulin-Resistant States and Decreases in FOXOs and PGC-1α and Increases in Aβ1-40/42 and Phospho-Tau May Abet Alzheimer Development. Diabetes 2016, 65, 1892–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, F.; Flavin, W.P.; Iqbal, S.; Pacelli, C.; Sri Renganathan, S.D.; Trudeau, L.E.; Campbell, E.M.; Fraser, P.E.; Tandon, A. Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J. Biol. Chem. 2016, 291, 4374–4385. [Google Scholar] [CrossRef] [Green Version]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Wang, M.W.; Sung, H.J.; Matoba, S.; Hwang, P.M. Polo-like kinases mediate cell survival in mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 14542–14546. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Deng, Y.; Qing, H. The phosphorylation of α-synuclein: Development and implication for the mechanism and therapy of the Parkinson’s disease. J. Neurochem. 2015, 135, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Inglis, K.J.; Chereau, D.; Brigham, E.F.; Chiou, S.S.; Schöbel, S.; Frigon, N.L.; Yu, M.; Caccavello, R.J.; Nelson, S.; Motter, R.; et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 2009, 284, 2598–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ma, W.; Wang, H.P.J.; Bunz, F.; Hwang, P.M. Polo-like kinase 2 activates an antioxidant pathway to promote the survival of cells with mitochondrial dysfunction. Free Radic. Biol. Med. 2014, 73, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Deng, X.; Shi, Y.; Su, Y.; Wei, J.; Duan, H. PGC-1alpha, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol. 2016, 229, R99–R115. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranin, E.; et al. Mitochondrial defect and PGC-1α dysfunction in parkin- associated familial Parkinson’s disease. BBA Mol. Basis Dis. 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.; Reddy, S.; Zheng, K.; Betensky, R.A.; Simon, D.K. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson’s disease. BMC Med. Genet. 2011, 12, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.-H.; Ko Han, S.; Kang, H.; Lee, Y.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1a contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Piqué, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.K.; Han, J.H.; Jang, J.Y.; Yun, S.E.; Kim, G.Y.; Kang, S.; Rhim, H. A novel link between the conformations, exposure of specific epitopes, and subcellular localization of α-synuclein. Biochim. Biophys. Acta 2015, 1850, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, C.-T.; Chen, K.-Y.; Wang, W.; Chiu, J.-Y.; Wu, D.; Chao, T.-Y.; Hu, C.-J.; Chau, K.-Y.D.; Bamodu, O.A. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020, 9, 740. https://doi.org/10.3390/cells9030740
Hong C-T, Chen K-Y, Wang W, Chiu J-Y, Wu D, Chao T-Y, Hu C-J, Chau K-YD, Bamodu OA. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells. 2020; 9(3):740. https://doi.org/10.3390/cells9030740
Chicago/Turabian StyleHong, Chien-Tai, Kai-Yun Chen, Weu Wang, Jing-Yuan Chiu, Dean Wu, Tsu-Yi Chao, Chaur-Jong Hu, Kai-Yin David Chau, and Oluwaseun Adebayo Bamodu. 2020. "Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling" Cells 9, no. 3: 740. https://doi.org/10.3390/cells9030740
APA StyleHong, C. -T., Chen, K. -Y., Wang, W., Chiu, J. -Y., Wu, D., Chao, T. -Y., Hu, C. -J., Chau, K. -Y. D., & Bamodu, O. A. (2020). Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells, 9(3), 740. https://doi.org/10.3390/cells9030740