Extracellular Vesicle Biomarkers Reveal Inhibition of Neuroinflammation by Infliximab in Association with Antidepressant Response in Adults with Bipolar Depression

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants
2.3. Procedures
2.4. Clinical Measures
2.5. Neuroimaging
2.6. Isolation of Extracellular Vesicles Enriched for Neuronal Origin
2.7. NEV Protein Quantification
2.8. Statistical Analysis
3. Results
3.1. Demographics and Clinical Characteristics
3.2. Baseline Associations between NEV Biomarkers and Clinical Variables
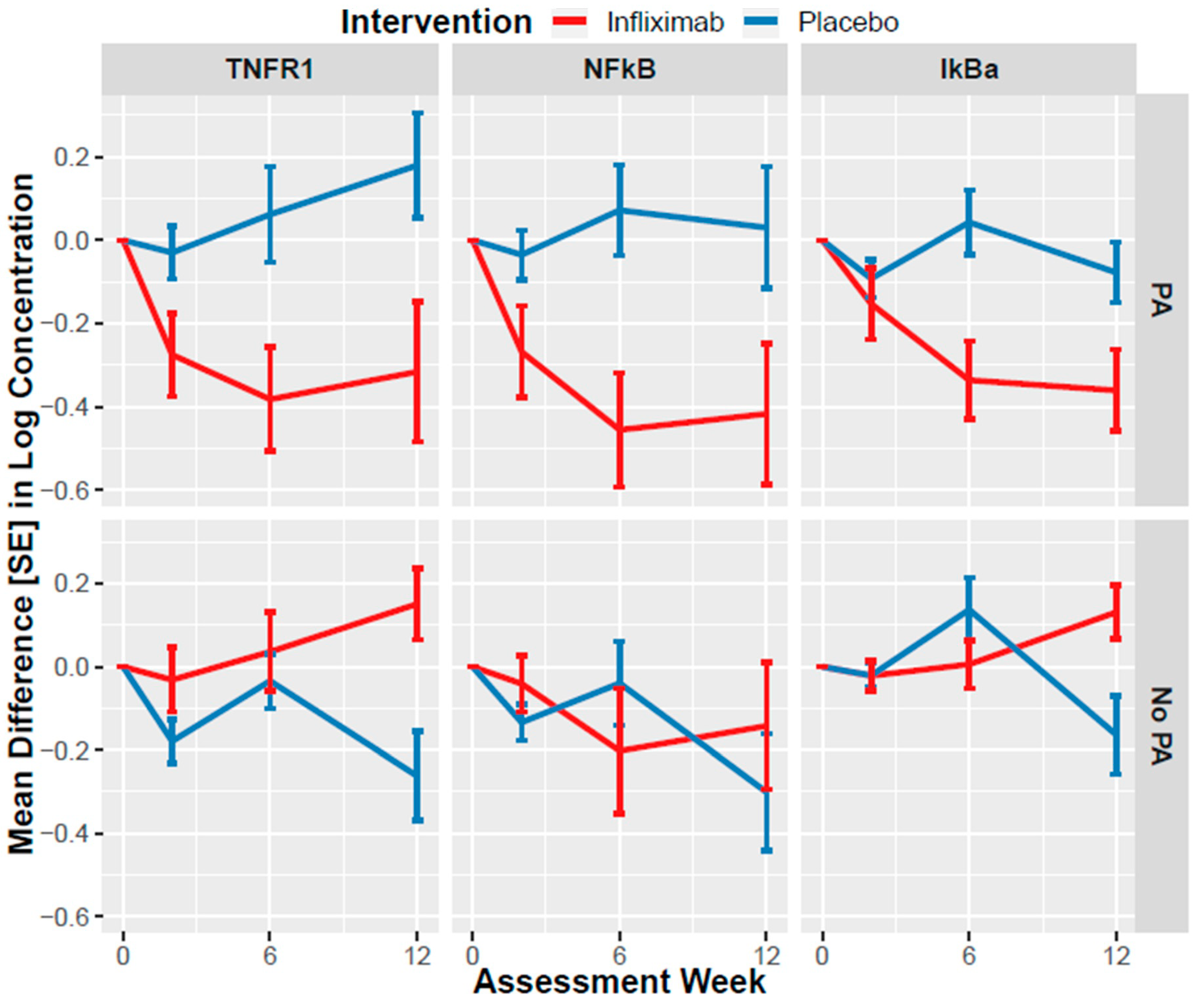
3.3. Association of Infliximab with NEV Biomarker Changes
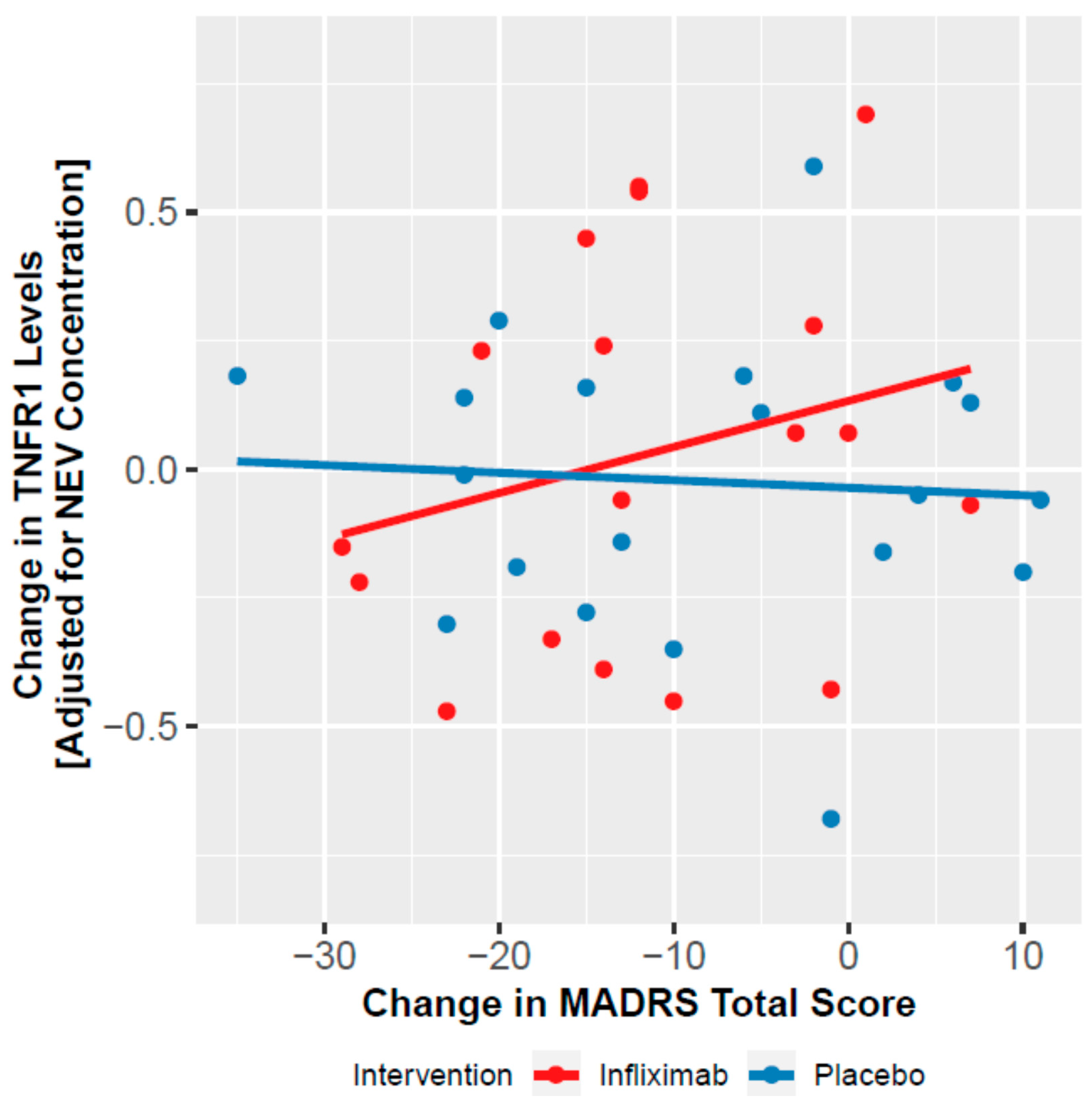
3.4. Moderation of Clinical Effect by NEV Biomarkers
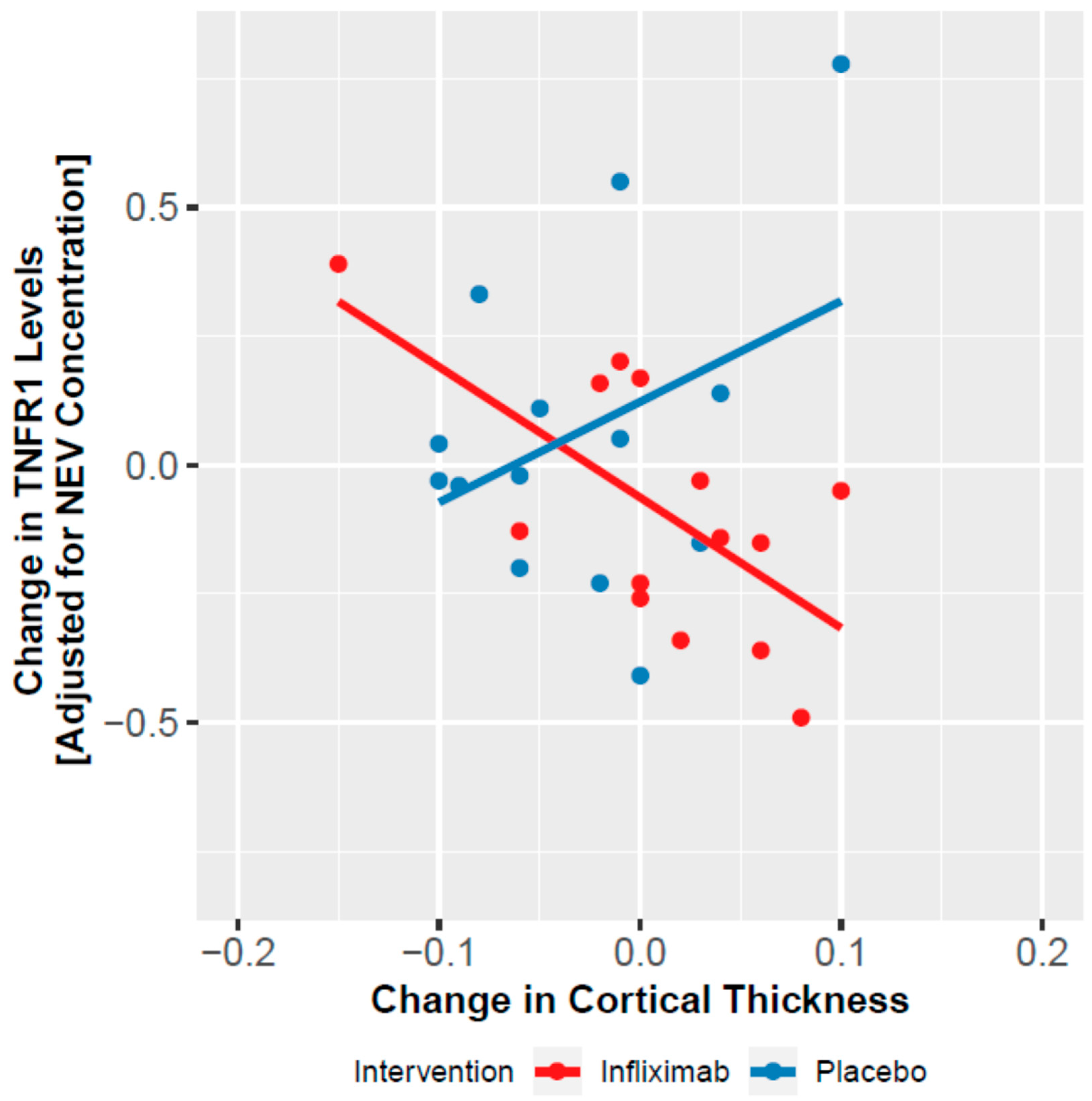
3.5. Change in NEV Biomarkers and Cortical Thickness
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Munkholm, K.; Brauner, J.V.; Kessing, L.V.; Vinberg, M. Cytokines in bipolar disorder vs. healthy control subjects: A systematic review and meta-analysis. J. Psychiatr. Res. 2013, 47, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Dargel, A.A.; Godin, O.; Kapczinski, F.; Kupfer, D.J.; Leboyer, M. C-reactive protein alterations in bipolar disorder: A meta-analysis. J. Clin. Psychiatry 2015, 76, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, V.V.; Sayana, P.; Pinjari, O.F.; Ahmad, N.; da Rosa, M.I.; Quevedo, J.; Barichello, T. Postmortem evidence of brain inflammatory markers in bipolar disorder: A systematic review. Mol. Psychiatry 2020, 25, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Haarman, B.C.; Riemersma-Van der Lek, R.F.; de Groot, J.C.; Ruhe, H.G.; Klein, H.C.; Zandstra, T.E.; Burger, H.; Schoevers, R.A.; de Vries, E.F.; Drexhage, H.A.; et al. Neuroinflammation in bipolar disorder—A [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav. Immun. 2014, 40, 219–225. [Google Scholar] [CrossRef]
- Wang, A.K.; Miller, B.J. Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder, and depression. Schizophr. Bull. 2018, 44, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Y.; Gildengers, A.G.; Hsu, J.L.; Chung, K.H.; Chen, P.H.; Huang, Y.J. Inflammation associated with volume reduction in the gray matter and hippocampus of older patients with bipolar disorder. J. Affect. Disord. 2019, 244, 60–66. [Google Scholar] [CrossRef]
- Lin, K.; Shao, R.; Wang, R.; Lu, W.; Zou, W.; Chen, K.; Gao, Y.; Brietzke, E.; McIntyre, R.S.; Mansur, R.B.; et al. Inflammation, brain structure and cognition interrelations among individuals with differential risks for bipolar disorder. Brain Behav. Immun. 2020, 83, 192–199. [Google Scholar] [CrossRef]
- Benedetti, F.; Poletti, S.; Hoogenboezem, T.A.; Mazza, E.; Ambree, O.; de Wit, H.; Wijkhuijs, A.J.; Locatelli, C.; Bollettini, I.; Colombo, C.; et al. Inflammatory cytokines influence measures of white matter integrity in Bipolar Disorder. J. Affect. Disord. 2016, 202, 1–9. [Google Scholar] [CrossRef]
- Chung, K.H.; Huang, S.H.; Wu, J.Y.; Chen, P.H.; Hsu, J.L.; Tsai, S.Y. The link between high-sensitivity C-reactive protein and orbitofrontal cortex in euthymic bipolar disorder. Neuropsychobiology 2013, 68, 168–173. [Google Scholar] [CrossRef]
- Savitz, J.; Dantzer, R.; Wurfel, B.E.; Victor, T.A.; Ford, B.N.; Bodurka, J.; Bellgowan, P.S.; Teague, T.K.; Drevets, W.C. Neuroprotective kynurenine metabolite indices are abnormally reduced and positively associated with hippocampal and amygdalar volume in bipolar disorder. Psychoneuroendocrinology 2015, 52, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Husain, M.I.; Strawbridge, R.; Stokes, P.R.; Young, A.H. Anti-inflammatory treatments for mood disorders: Systematic review and meta-analysis. J. Psychopharmacol. 2017, 31, 1137–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblat, J.D.; Kakar, R.; Berk, M.; Kessing, L.V.; Vinberg, M.; Baune, B.T.; Mansur, R.B.; Brietzke, E.; Goldstein, B.I.; McIntyre, R.S. Anti-inflammatory agents in the treatment of bipolar depression: A systematic review and meta-analysis. Bipolar Disord. 2016, 18, 89–101. [Google Scholar] [CrossRef]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.B.; Khandaker, G.M. Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 2018, 23, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Subramaniapillai, M.; Lee, Y.; Pan, Z.; Carmona, N.E.; Shekotikhina, M.; Rosenblat, J.D.; Brietzke, E.; Soczynska, J.K.; Cosgrove, V.E.; et al. Efficacy of adjunctive infliximab vs placebo in the treatment of adults with bipolar I/II depression: A randomized clinical trial. JAMA Psychiatry 2019, 76, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Raison, C.L.; Woolwine, B.J.; Haroon, E.; Binder, E.B.; Miller, A.H.; Felger, J.C. Transcriptional signatures related to glucose and lipid metabolism predict treatment response to the tumor necrosis factor antagonist infliximab in patients with treatment-resistant depression. Brain Behav. Immun. 2013, 31, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, D.; Akhtar, R.; Ciufolini, S.; Pariante, C.M.; Mondelli, V. Childhood trauma and adulthood inflammation: A meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-alpha. Mol. Psychiatry 2016, 21, 642–649. [Google Scholar] [CrossRef] [Green Version]
- Coelho, R.; Viola, T.W.; Walss-Bass, C.; Brietzke, E.; Grassi-Oliveira, R. Childhood maltreatment and inflammatory markers: A systematic review. Acta Psychiatr. Scand. 2014, 129, 180–192. [Google Scholar] [CrossRef]
- Pulliam, L.; Sun, B.; Mustapic, M.; Chawla, S.; Kapogiannis, D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J. Neurovirol. 2019, 25, 702–709. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of extracellular vesicle biomarkers with alzheimer disease in the baltimore longitudinal study of aging. JAMA Neurol. 2019, 76, 420–429. [Google Scholar] [CrossRef]
- Athauda, D.; Gulyani, S.; Karnati, H.; Li, Y.; Tweedie, D.; Mustapic, M.; Chawla, S.; Chowdhury, K.; Skene, S.S.; Greig, N.H.; et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with parkinson disease: A secondary analysis of the exenatide-PD trial. JAMA Neurol. 2019, 76, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Mustapic, M.; Eitan, E.; Werner, J.K., Jr.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma extracellular vesicles enriched for neuronal origin: A potential window into brain pathologic processes. Front. Neurosci. 2017, 11, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Ledreux, A.; Granholm, A.C.; Elahi, F.M.; Goetzl, L.; Hiramoto, J.; Kapogiannis, D. Neuron-derived exosome proteins may contribute to progression from repetitive mild traumatic brain injuries to chronic traumatic encephalopathy. Front. Neurosci. 2019, 13, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, J.; Mustapic, M.; Diaz-Arrastia, R.; Lange, R.; Gulyani, S.; Diehl, T.; Motamedi, V.; Osier, N.; Stern, R.A.; Kapogiannis, D. Higher exosomal tau, amyloid-beta 42 and IL-10 are associated with mild TBIs and chronic symptoms in military personnel. Brain Injury 2018, 32, 1277–1284. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Wijtenburg, S.A.; Kapogiannis, D.; Korenic, S.A.; Mullins, R.J.; Tran, J.; Gaston, F.E.; Chen, S.; Mustapic, M.; Hong, L.E.; Rowland, L.M. Brain insulin resistance and altered brain glucose are related to memory impairments in schizophrenia. Schizophr. Res. 2019, 208, 324–330. [Google Scholar] [CrossRef]
- Mustapic, M.; Tran, J.; Craft, S.; Kapogiannis, D. Extracellular vesicle biomarkers track cognitive changes following intranasal insulin in Alzheimer’s disease. J. Alzheimers Dis. 2019, 69, 489–498. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular vesicle-associated abeta mediates trans-neuronal bioenergetic and Ca(2+)-handling deficits in Alzheimer’s disease models. NPJ Aging Mech. Dis. 2016, 2. [Google Scholar] [CrossRef]
- Torices, S.; Julia, A.; Munoz, P.; Varela, I.; Balsa, A.; Marsal, S.; Fernandez-Nebro, A.; Blanco, F.; Lopez-Hoyos, M.; Martinez-Taboada, V.; et al. A functional variant of TLR10 modifies the activity of NFkB and may help predict a worse prognosis in patients with rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 221. [Google Scholar] [CrossRef] [PubMed]
- Kokolakis, G.; Giannikaki, E.; Stathopoulos, E.; Avramidis, G.; Tosca, A.D.; Kruger-Krasagakis, S. Infliximab restores the balance between pro- and anti-apoptotic proteins in regressing psoriatic lesions. Br. J. Dermatol. 2012, 166, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Eggert, M.; Seeck, U.; Semmler, M.; Maass, U.; Dietmann, S.; Schulz, M.; Dotzlaw, H.; Neeck, G. An evaluation of anti-TNF-alpha-therapy in patients with ankylosing spondylitis: Imbalanced activation of NF kappa B subunits in lymphocytes and modulation of serum cortisol concentration. Rheumatol. Int. 2007, 27, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Dale, A.M.; Fischl, B.; Sereno, M.I. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage 1999, 9, 179–194. [Google Scholar] [CrossRef]
- Fischl, B.; Sereno, M.I.; Dale, A.M. Cortical surface-based analysis. II. Inflation, flattening, and a surface-based coordinate system. Neuroimage 1999, 9, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Reuter, M.; Schmansky, N.J.; Rosas, H.D.; Fischl, B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 2012, 61, 1402–1418. [Google Scholar] [CrossRef] [Green Version]
- Fischl, B.; Dale, A.M. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc. Natl. Acad. Sci. USA 2000, 97, 11050–11055. [Google Scholar] [CrossRef] [Green Version]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-‘t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological guidelines to study extracellular vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Agliardi, C.; Guerini, F.R.; Zanzottera, M.; Bianchi, A.; Nemni, R.; Clerici, M. SNAP-25 in serum is carried by exosomes of neuronal origin and is a potential biomarker of Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 5792–5798. [Google Scholar] [CrossRef]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimers Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the assessment of Abeta42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakim, M.S.; Ding, S.; Chen, S.; Yin, Y.; Su, J.; van der Woude, C.J.; Fuhler, G.M.; Peppelenbosch, M.P.; Pan, Q.; Wang, W. TNF-alpha exerts potent anti-rotavirus effects via the activation of classical NF-kappaB pathway. Virus Res. 2018, 253, 28–37. [Google Scholar] [CrossRef]
- Samidurai, M.; Ramasamy, V.S.; Jo, J. beta-amyloid inhibits hippocampal LTP through TNFR/IKK/NF-kappaB pathway. Neurol. Res. 2018, 40, 268–276. [Google Scholar] [CrossRef]
- Chengke, L.; Weiwei, L.; Xiyang, W.; Ping, W.; Xiaoyang, P.; Zhengquan, X.; Hao, Z.; Penghui, Z.; Wei, P. Effect of infliximab combined with methylprednisolone on expressions of NF-kappaB, TRADD, and FADD in rat acute spinal cord injury. Spine 2013, 38, E861–E869. [Google Scholar] [CrossRef]
- Mahmoud, M.F.; El Shazly, S.M.; Barakat, W. Inhibition of TNF-alpha protects against hepatic ischemia-reperfusion injury in rats via NF-kappaB dependent pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 465–471. [Google Scholar] [CrossRef]
- Kanbe, K.; Inoue, K.; Inoue, Y.; Suzuki, Y. Histological analysis of synovium in cases of effect attenuation associated with infliximab therapy in rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 777–781. [Google Scholar] [CrossRef]
- Dadsetan, S.; Balzano, T.; Forteza, J.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Hernandez-Rabaza, V.; Gil-Perotin, S.; Cubas-Nunez, L.; Garcia-Verdugo, J.M.; Agusti, A.; et al. Reducing peripheral inflammation with infliximab reduces neuroinflammation and improves cognition in rats with hepatic encephalopathy. Front. Mol. Neurosci. 2016, 9, 106. [Google Scholar] [CrossRef]
- Dadsetan, S.; Balzano, T.; Forteza, J.; Agusti, A.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Hernandez-Rabaza, V.; Gomez-Gimenez, B.; ElMlili, N.; Llansola, M.; et al. Infliximab reduces peripheral inflammation, neuroinflammation, and extracellular GABA in the cerebellum and improves learning and motor coordination in rats with hepatic encephalopathy. J. Neuroinflamm. 2016, 13, 245. [Google Scholar] [CrossRef] [Green Version]
- Pace, T.W.; Mletzko, T.C.; Alagbe, O.; Musselman, D.L.; Nemeroff, C.B.; Miller, A.H.; Heim, C.M. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am. J. Psychiatry 2006, 163, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.; Wingenfeld, K.; Schmidt, I.; Meinlschmidt, G.; Hellhammer, D.H.; Heim, C.M. Increased peripheral NF-kappaB pathway activity in women with childhood abuse-related posttraumatic stress disorder. Brain Behav. Immun. 2012, 26, 13–17. [Google Scholar] [CrossRef] [PubMed]
- do Prado, C.H.; Grassi-Oliveira, R.; Daruy-Filho, L.; Wieck, A.; Bauer, M.E. Evidence for immune activation and resistance to glucocorticoids following childhood maltreatment in adolescents without psychopathology. Neuropsychopharmacology 2017, 42, 2272–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklowitz, D.J.; Portnoff, L.C.; Armstrong, C.C.; Keenan-Miller, D.; Breen, E.C.; Muscatell, K.A.; Eisenberger, N.I.; Irwin, M.R. Inflammatory cytokines and nuclear factor-kappa B activation in adolescents with bipolar and major depressive disorders. Psychiatry Res. 2016, 241, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.M.; Su, T.P.; Li, C.T.; Tsai, S.J.; Chen, M.H.; Tu, P.C.; Chiou, W.F. Comparison of pro-inflammatory cytokines among patients with bipolar disorder and unipolar depression and normal controls. Bipolar Disord. 2015, 17, 269–277. [Google Scholar] [CrossRef]
- Doganavsargil-Baysal, O.; Cinemre, B.; Aksoy, U.M.; Akbas, H.; Metin, O.; Fettahoglu, C.; Gokmen, Z.; Davran, F. Levels of TNF-alpha, soluble TNF receptors (sTNFR1, sTNFR2), and cognition in bipolar disorder. Hum. Psychopharmacol. 2013, 28, 160–167. [Google Scholar] [CrossRef]
- Hoseth, E.Z.; Ueland, T.; Dieset, I.; Birnbaum, R.; Shin, J.H.; Kleinman, J.E.; Hyde, T.M.; Morch, R.H.; Hope, S.; Lekva, T.; et al. A study of TNF pathway activation in schizophrenia and bipolar disorder in plasma and brain tissue. Schizophr. Bull. 2017, 43, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, A.L.; de Sousa, R.T.; Zanetti, M.V.; Brunoni, A.R.; Busatto, G.F.; Zarate, C.A., Jr.; Gattaz, W.F.; Machado-Vieira, R. Increased plasma levels of soluble TNF receptors 1 and 2 in bipolar depression and impact of lithium treatment. Hum. Psychopharmacol. 2015, 30, 52–56. [Google Scholar] [CrossRef]
- Munkholm, K.; Vinberg, M.; Vedel Kessing, L. Cytokines in bipolar disorder: A systematic review and meta-analysis. J. Affect. Disord. 2013, 144, 16–27. [Google Scholar] [CrossRef]
- Grassi-Oliveira, R.; Brietzke, E.; Pezzi, J.C.; Lopes, R.P.; Teixeira, A.L.; Bauer, M.E. Increased soluble tumor necrosis factor-alpha receptors in patients with major depressive disorder. Psychiatry Clin. Neurosci. 2009, 63, 202–208. [Google Scholar] [CrossRef]
- Huang, M.H.; Chen, M.H.; Tu, P.C.; Bai, Y.M.; Su, T.P.; Yang, B.H.; Liu, R.S.; Li, C.T. Elevated tumor necrosis factor-alpha receptor subtype 1 and the association with abnormal brain function in treatment-resistant depression. J. Affect. Disord. 2018, 235, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Dellarole, A.; Morton, P.; Brambilla, R.; Walters, W.; Summers, S.; Bernardes, D.; Grilli, M.; Bethea, J.R. Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav. Immun. 2014, 41, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguz-Lecznar, M.; Zakrzewska, R.; Kossut, M. Inhibition of Tnf-alpha R1 signaling can rescue functional cortical plasticity impaired in early post-stroke period. Neurobiol. Aging 2015, 36, 2877–2884. [Google Scholar] [CrossRef]
- Becker, D.; Deller, T.; Vlachos, A. Tumor necrosis factor (TNF)-receptor 1 and 2 mediate homeostatic synaptic plasticity of denervated mouse dentate granule cells. Sci. Rep. 2015, 5, 12726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1beta Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Luo, Y.; He, Y.; Zhang, H.; Zhang, R.; Li, X.; Dobrucki, W.L.; Sinusas, A.J.; Sessa, W.C.; Min, W. Differential functions of tumor necrosis factor receptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angiogenesis. Am. J. Pathol. 2006, 169, 1886–1898. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.A.; Weber, R.F.; Figari, I.S.; Reynolds, C.; Palladino, M.A., Jr.; Goeddel, D.V. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. USA 1991, 88, 9292–9296. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Nanes, M.S. Tumor necrosis factor-alpha: Molecular and cellular mechanisms in skeletal pathology. Gene 2003, 321, 1–15. [Google Scholar] [CrossRef]
- MacEwan, D.J. TNF receptor subtype signalling: Differences and cellular consequences. Cell Signal. 2002, 14, 477–492. [Google Scholar] [CrossRef]
- Russell, A.E.; Jun, S.; Sarkar, S.; Geldenhuys, W.J.; Lewis, S.E.; Rellick, S.L.; Simpkins, J.W. Extracellular vesicles secreted in response to cytokine exposure increase mitochondrial oxygen consumption in recipient cells. Front. Cell. Neurosci. 2019, 13, 51. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | Placebo (n = 28) | Infliximab (n = 27) | p-Value |
|---|---|---|---|
| Age (years), mean (SD) | 45.75 (10.28) | 44.04 (11.55) | 0.564 a |
| Gender (female), n (%) | 24 (85.7) | 20 (74.1) | 0.281 b |
| Ethnicity (Caucasian), n (%) | 5 (17.9) | 4 (14.8) | 0.760 b |
| Education, n (%) | |||
| High school | 4 (14.8) | 5 (19.2) | 0.282 b |
| College/University | 22 (81.5) | 17 (65.4) | |
| Graduate school | 1 (3.7) | 4 (15.4) | |
| MADRS (total score), mean (SD) | 30.07 (6.72) | 31.33 (6.85) | 0.595 c |
| YMRS (total score), mean (SD) | 4.71 (4.30) | 3.48 (3.08) | 0.377 c |
| BMI (kg/m2), mean (SD) | 34.55 (7.66) | 34.57 (10.08) | 0.608 c |
| Tobacco use, n (%) | 8 (28.6) | 10 (37.0) | 0.504 b |
| Age at onset (years), mean (SD) | 17.14 (9.32) | 18.59 (8.13) | 0.437 c |
| Number of lifetime psychiatric hospitalizations, mean (SD) | 1.58 (2.02) | 1.74 (1.91) | 0.679 c |
| Length of current depressive episode (months), mean (SD) | 11.67 (20.80) | 11.85 (15.48) | 0.378 c |
| Medications | |||
| Antipsychotic, n (%) | 16 (66.7) | 18 (69.2) | 0.846 b |
| Antidepressant, n (%) | 18 (75.0) | 15 (57.7) | 0.197 b |
| Lithium, n (%) | 5 (20.8) | 6 (22.20 | 0.904 b |
| Anticonvulsants, n (%) | 10 (41.7) | 15 (57.7) | 0.258 b |
| Antidiabetic, n (%) | 4 (14.3) | 4 (14.8) | 0.956 b |
| Childhood Trauma Questionnaire (total score), mean (SD) | 54.46 (17.81) | 56.51 (22.54) | 0.866 c |
| Physical Abuse, mean (SD) | 9.35 (5.61) | 8.48 (5.67) | 0.266 c |
| NEV | MADRS | CRP | CTQ Total Score | |||
|---|---|---|---|---|---|---|
| RR | p-Value a | RR | p-Value b | RR | p-Value c | |
| TNFR1 | 0.944 | 0.014 | 1.176 | 0.026 | 1.002 | 0.955 |
| TNFR2 | 1.097 | 0.110 | 2.021 | <0.001 | 1.190 | 0.072 |
| NF-κB | 0.954 | 0.015 | 1.166 | 0.011 | 1.047 | 0.156 |
| c-Myc | 0.961 | 0.100 | 1.178 | 0.026 | 1.042 | 0.288 |
| FADD | 0.962 | 0.098 | 1.217 | 0.007 | 1.067 | 0.094 |
| IKKα/β | 0.965 | 0.226 | 1.385 | <0.001 | 1.048 | 0.346 |
| IκBα | 0.941 | 0.056 | 1.389 | <0.001 | 1.080 | 0.136 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansur, R.B.; Delgado-Peraza, F.; Subramaniapillai, M.; Lee, Y.; Iacobucci, M.; Rodrigues, N.; Rosenblat, J.D.; Brietzke, E.; Cosgrove, V.E.; Kramer, N.E.; et al. Extracellular Vesicle Biomarkers Reveal Inhibition of Neuroinflammation by Infliximab in Association with Antidepressant Response in Adults with Bipolar Depression. Cells 2020, 9, 895. https://doi.org/10.3390/cells9040895
Mansur RB, Delgado-Peraza F, Subramaniapillai M, Lee Y, Iacobucci M, Rodrigues N, Rosenblat JD, Brietzke E, Cosgrove VE, Kramer NE, et al. Extracellular Vesicle Biomarkers Reveal Inhibition of Neuroinflammation by Infliximab in Association with Antidepressant Response in Adults with Bipolar Depression. Cells. 2020; 9(4):895. https://doi.org/10.3390/cells9040895
Chicago/Turabian StyleMansur, Rodrigo B., Francheska Delgado-Peraza, Mehala Subramaniapillai, Yena Lee, Michelle Iacobucci, Nelson Rodrigues, Joshua D. Rosenblat, Elisa Brietzke, Victoria E. Cosgrove, Nicole E. Kramer, and et al. 2020. "Extracellular Vesicle Biomarkers Reveal Inhibition of Neuroinflammation by Infliximab in Association with Antidepressant Response in Adults with Bipolar Depression" Cells 9, no. 4: 895. https://doi.org/10.3390/cells9040895
APA StyleMansur, R. B., Delgado-Peraza, F., Subramaniapillai, M., Lee, Y., Iacobucci, M., Rodrigues, N., Rosenblat, J. D., Brietzke, E., Cosgrove, V. E., Kramer, N. E., Suppes, T., Raison, C. L., Chawla, S., Nogueras-Ortiz, C., McIntyre, R. S., & Kapogiannis, D. (2020). Extracellular Vesicle Biomarkers Reveal Inhibition of Neuroinflammation by Infliximab in Association with Antidepressant Response in Adults with Bipolar Depression. Cells, 9(4), 895. https://doi.org/10.3390/cells9040895