CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus

Abstract
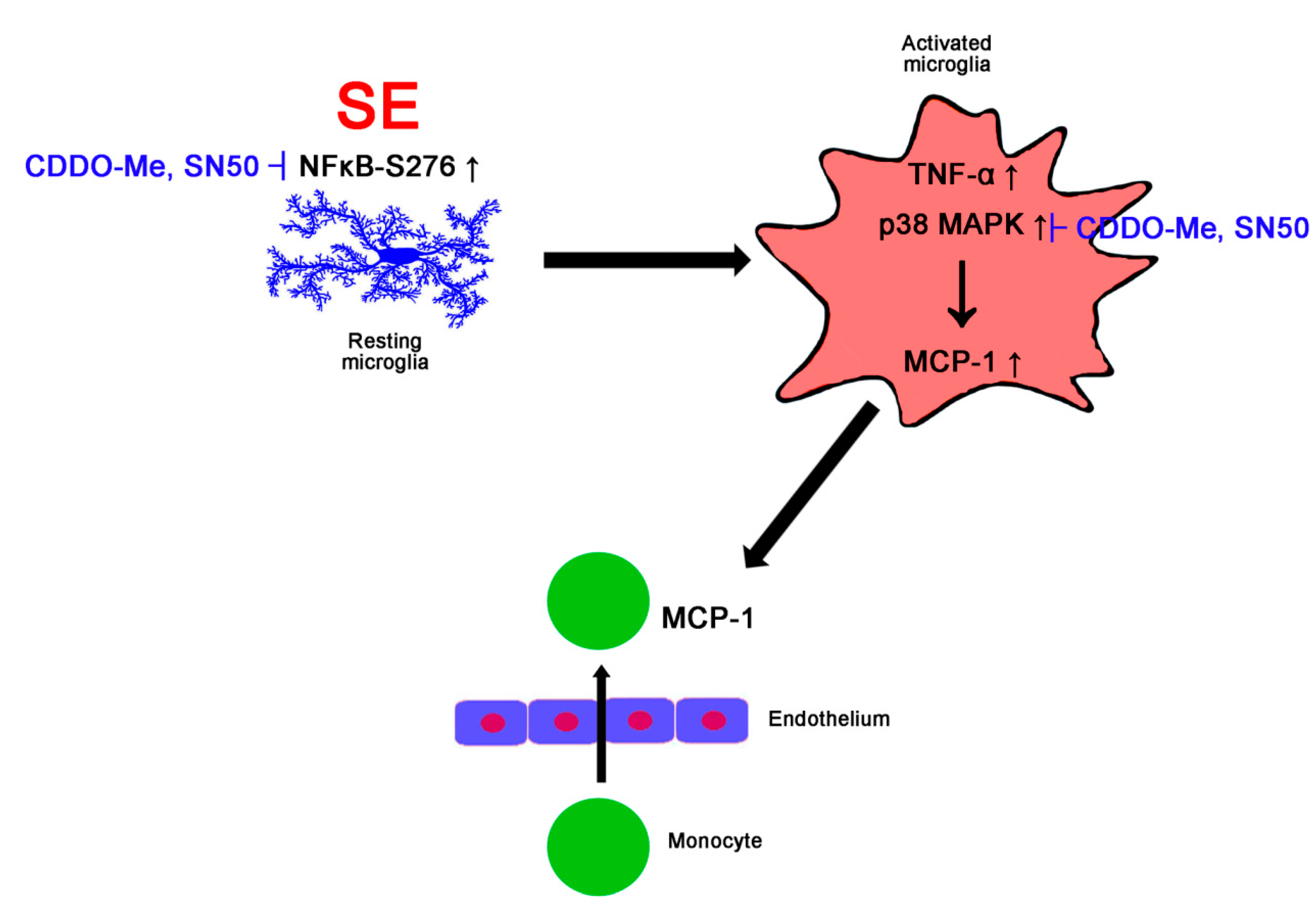
:1. Introduction
2. Materials and Method
2.1. Experimental Animals and Chemicals
2.2. Surgery and Drug Infusion
2.3. SE Induction
2.4. Tissue Processing and Immunohistochemistry
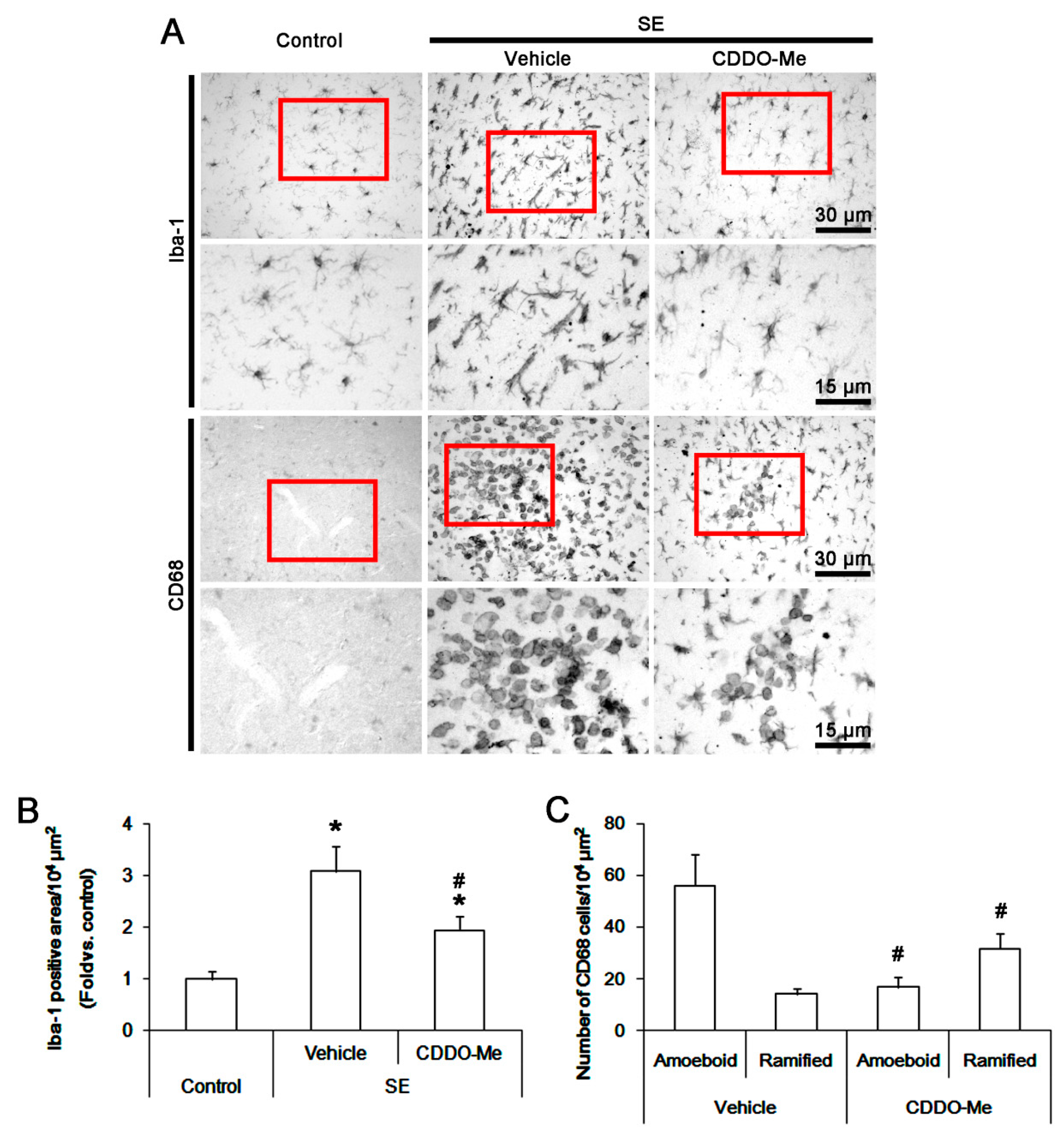
2.5. Cell Count and Measurements of Iba-1 Positive Area and Fluorescent Intensity
2.6. Western Blot
2.7. Data Analysis
3. Results
3.1. CDDO-Me Influences Monocyte Infiltration and Microglial Morphogenesis Induced by SE
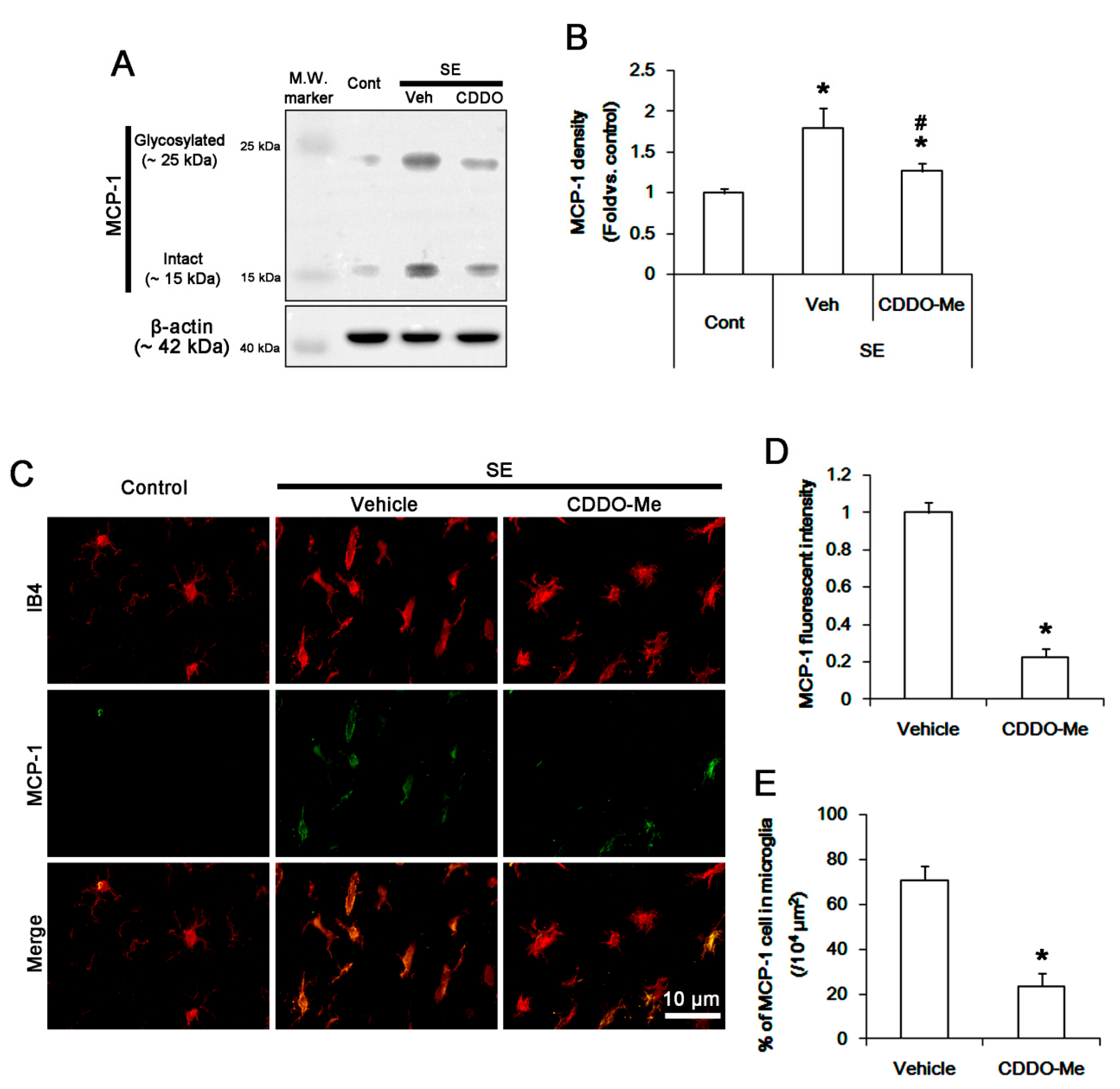
3.2. CDDO-Me Mitigated Monocyte Infiltration by Inhibiting Microglial MCP-1 Production Following SE
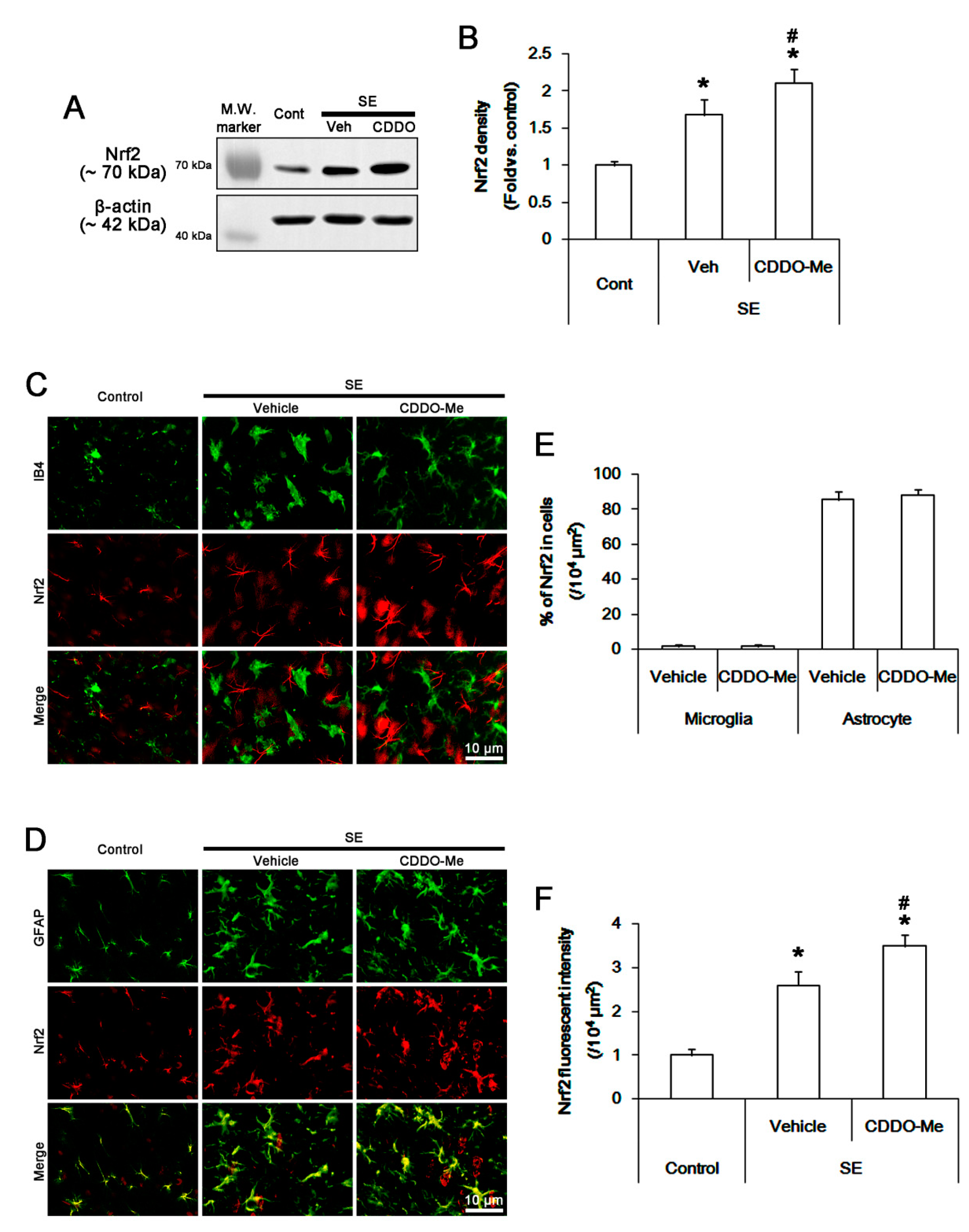
3.3. CDDO-Me Abolishes Microglial MCP-1 Production Independent of Nrf2 Activation Following SE
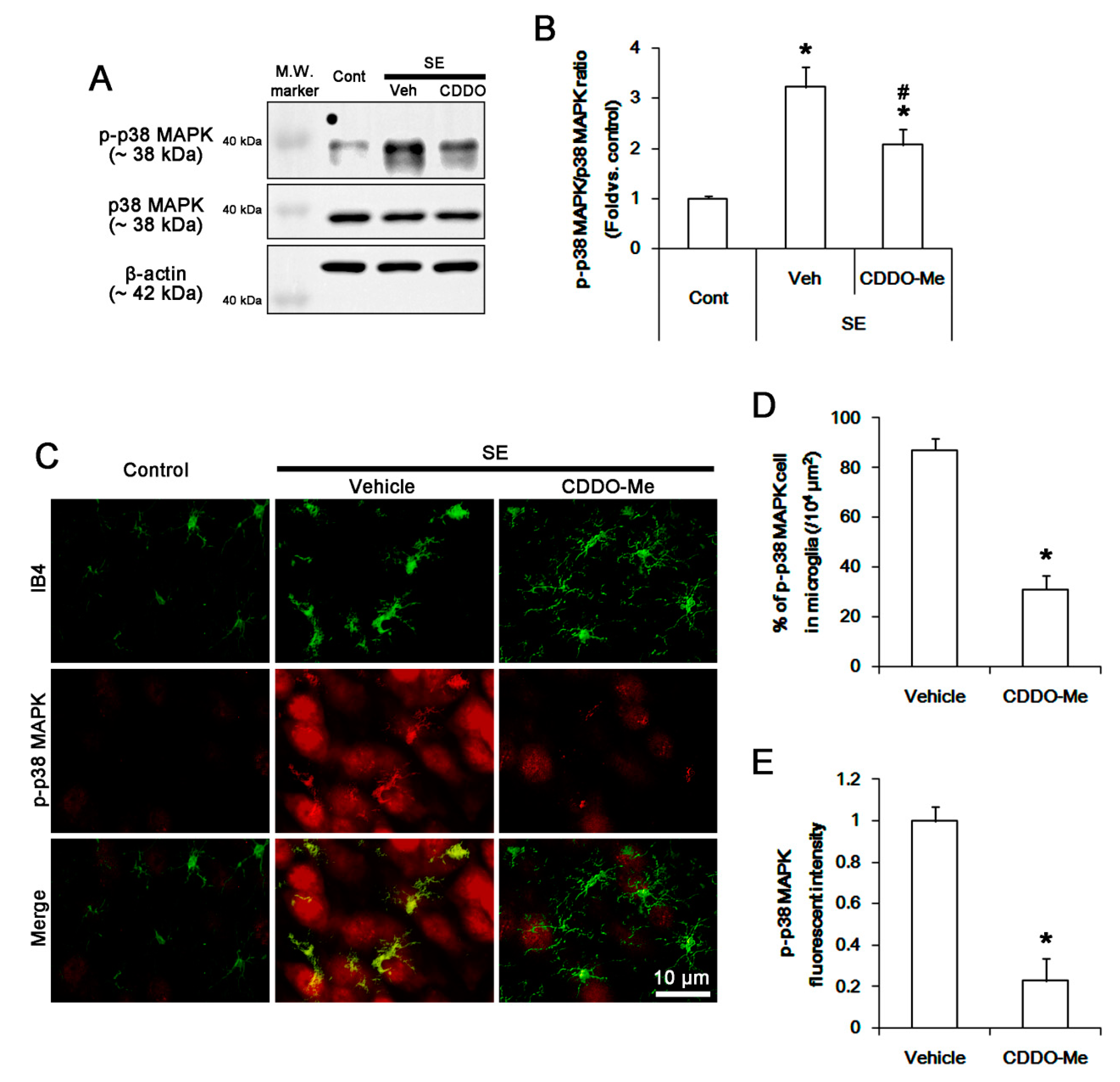
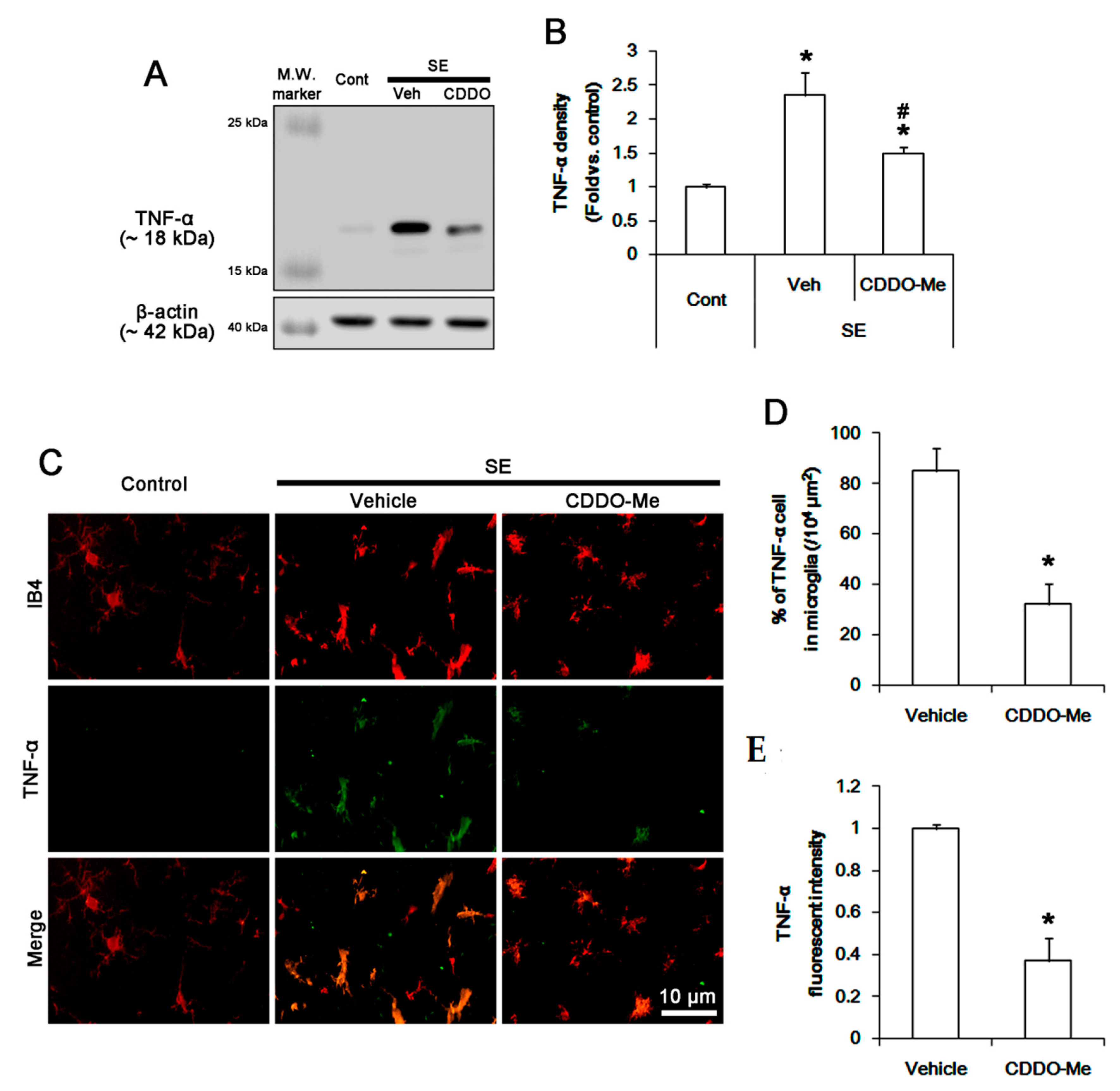
3.4. CDDO-Me Inhibits P38 MAPK Phosphorylation in Microglia Following SE
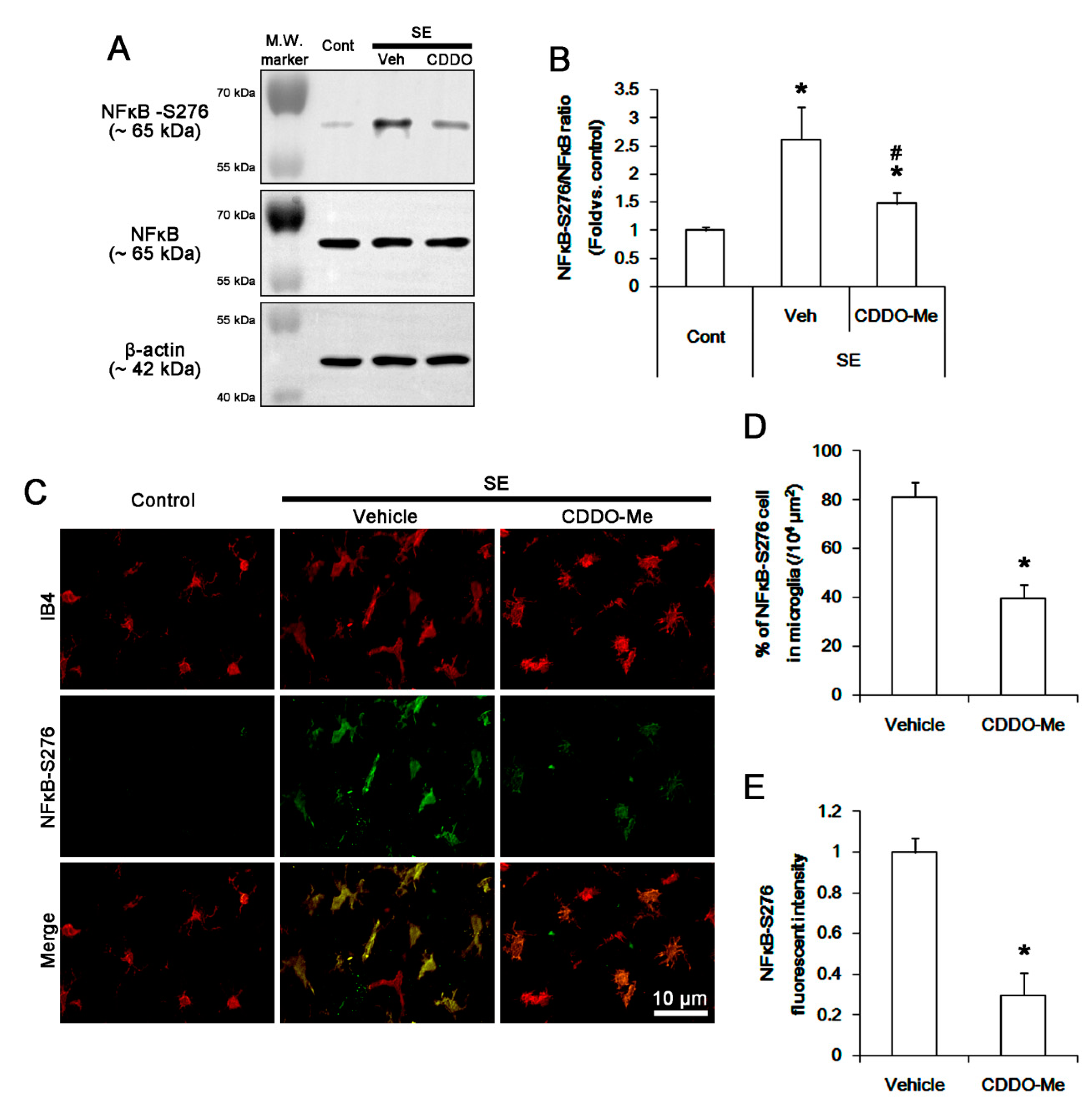
3.5. CDDO-Me Inhibits NFκB-S276 Phosphorylation in Microglia Following SE
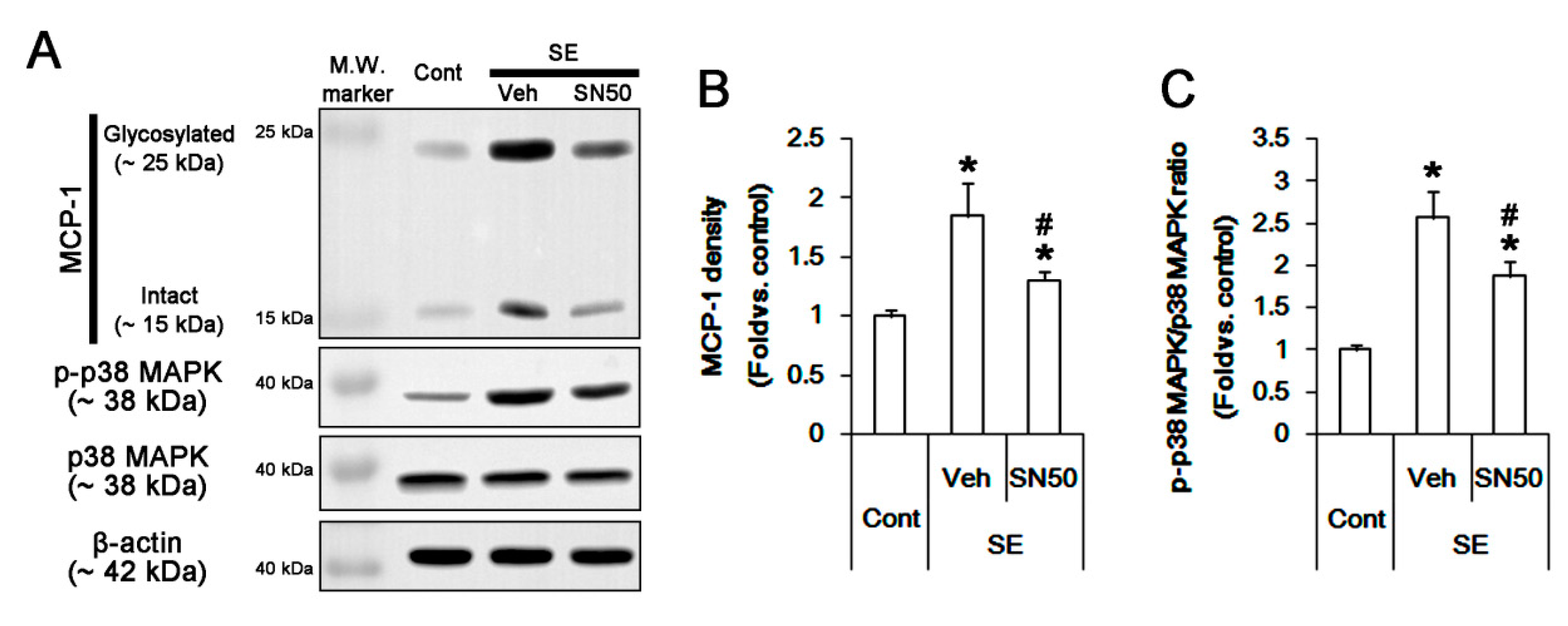
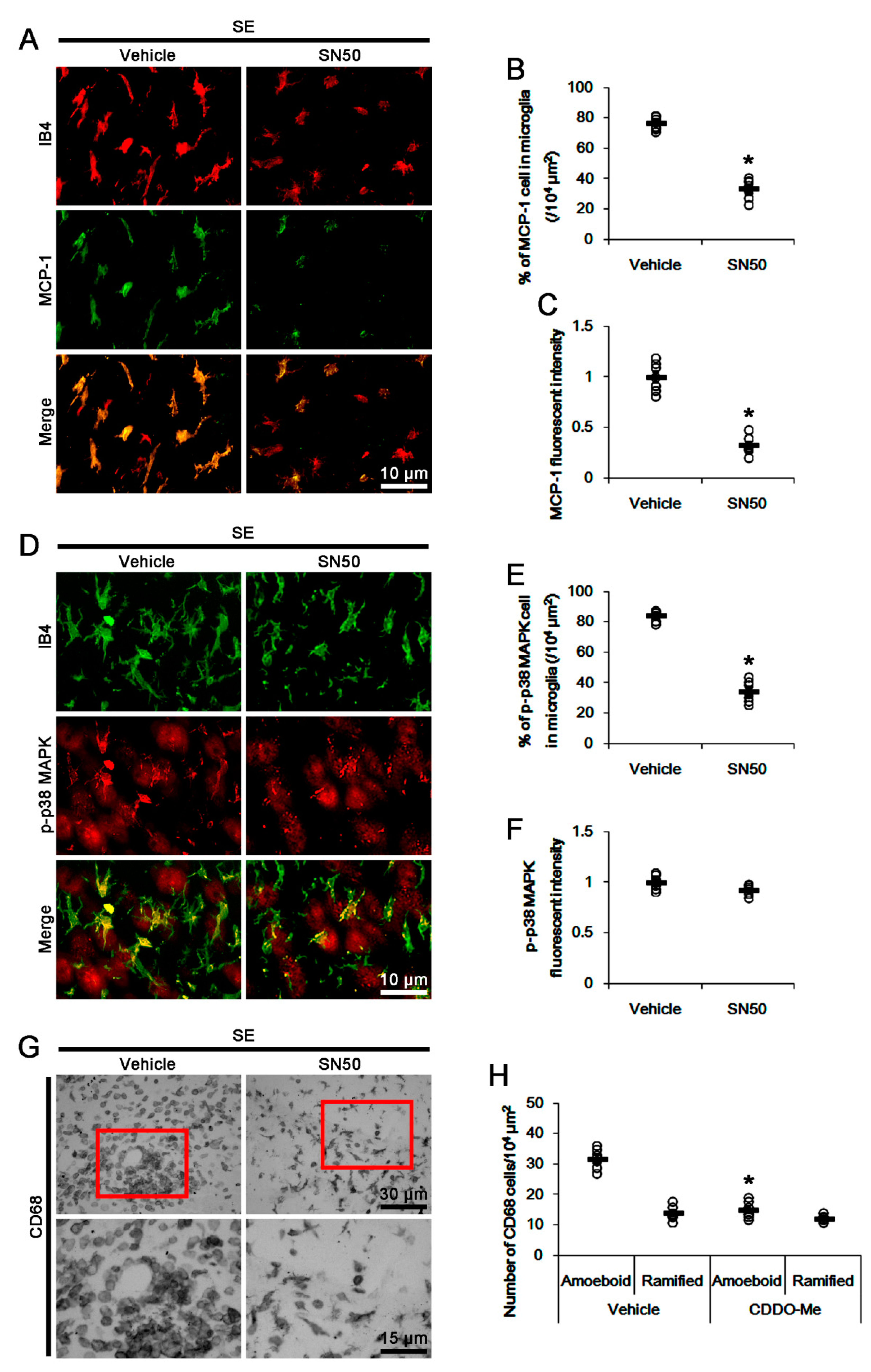
3.6. SN50 Mitigates MCP-1-Mediated Monocyte Infiltration Following SE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Katsumoto, A.; Lu, H.; Miranda, A.S.; Ransohoff, R.M. Ontogeny and functions of central nervous system macrophages. J. Immunol. 2014, 193, 2615–2621. [Google Scholar] [CrossRef]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef]
- Quagliato, L.A.; Nardi, A.E. The role of convergent ion channel pathways in microglial phenotypes: A systematic review of the implications for neurological and psychiatric disorders. Transl. Psychiatry 2018, 8, 259. [Google Scholar] [CrossRef]
- De Simoni, M.G.; Perego, C.; Ravizza, T.; Moneta, D.; Conti, M.; Marchesi, F.; De Luigi, A.; Garattini, S.; Vezzani, A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 2000, 12, 2623–2633. [Google Scholar] [CrossRef]
- Plata-Salamán, C.R.; Ilyin, S.E.; Turrin, N.P.; Gayle, D.; Flynn, M.C.; Romanovitch, A.E.; Kelly, M.E.; Bureau, Y.; Anisman, H.; McIntyre, D.C. Kindling modulates the IL-1beta system, TNF-alpha, TGF-beta1, and neuropeptide mRNAs in specific brain regions. Brain Res. Mol. Brain Res. 2000, 75, 248–258. [Google Scholar] [CrossRef]
- Rizzi, M.; Perego, C.; Aliprandi, M.; Richichi, C.; Ravizza, T.; Colella, D.; Velískŏvá, J.; Moshé, S.L.; De Simoni, M.G.; Vezzani, A. Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol. Dis. 2003, 14, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Andoh, M.; Ikegaya, Y.; Koyama, R. Synaptic Pruning by Microglia in Epilepsy. J. Clin. Med. 2019, 8, 2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Kang, T.C. P2X7 receptor regulates leukocyte infiltrations in rat frontoparietal cortex following status epilepticus. J. Neuroinflammation 2010, 7, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, M.W.; Roth, S.J.; Luther, E.; Rose, S.S.; Springer, T.A. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc. Natl. Acad. Sci. USA 1994, 91, 3652–3656. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, M.E.; Durham, S.K.; Swerdel, M.R.; Lewin, A.C.; Barton, D.S.; Megill, J.R.; Bravo, R.; Lira, S.A. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J. Immunol. 1995, 155, 5769–5776. [Google Scholar]
- Dinkel, K.; Dhabhar, F.S.; Sapolsky, R.M. Neurotoxic effects of polymorphonuclear granulocytes on hippocampal primary cultures. Proc. Natl. Acad. Sci. USA 2004, 101, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T.; Barry, B.; Hickey, W.F. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J. Immunol. 2001, 166, 4634–4643. [Google Scholar] [CrossRef] [Green Version]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Hiragi, T.; Ikegaya, Y.; Koyama, R. Microglia after Seizures and in Epilepsy. Cells 2018, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Taniguchi, R.; Matsuo, T.; Oguro, A.; Vogel, C.F.A.; Yamazaki, T.; Ishihara, Y. Suppressive effects of levetiracetam on neuroinflammation and phagocytic microglia: A comparative study of levetiracetam, valproate and carbamazepine. Neurosci. Lett. 2019, 708, 134363. [Google Scholar] [CrossRef]
- Hossain, M.M.; Weig, B.; Reuhl, K.; Gearing, M.; Wu, L.J.; Richardson, J.R. The anti-parkinsonian drug zonisamide reduces neuroinflammation: Role of microglial Na(v) 1.6. Exp. Neurol. 2018, 308, 111–119. [Google Scholar] [CrossRef]
- Chen, S.; Ye, J.; Chen, X.; Shi, J.; Wu, W.; Lin, W.; Lin, W.; Li, Y.; Fu, H.; Li, S. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-κB pathway dependent of HDAC3. J. Neuroinflammation 2018, 15, 150. [Google Scholar] [CrossRef]
- Rosa, A.S.; Freitas, M.F.; Rocha, I.R.; Chacur, M. Gabapentin decreases microglial cells and reverses bilateral hyperalgesia and allodynia in rats with chronic myositis. Eur. J. Pharmacol. 2017, 799, 111–117. [Google Scholar] [CrossRef]
- Chuang, Y.F.; Yang, H.Y.; Ko, T.L.; Hsu, Y.F.; Sheu, J.R.; Ou, G.; Hsu, M.J. Valproic acid suppresses lipopolysaccharide-induced cyclooxygenase-2 expression via MKP-1 in murine brain microvascular endothelial cells. Biochem. Pharmacol. 2014, 88, 372–383. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, H.C.; Song, H.K.; Jo, S.M.; Kim, D.S.; Choi, S.Y.; Kim, Y.I.; Kang, T.C. Levetiracetam inhibits interleukin-1 beta inflammatory responses in the hippocampus and piriform cortex of epileptic rats. Neurosci. Lett. 2010, 471, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Motaghinejad, M.; Motevalian, M. Involvement of AMPA/kainate and GABAA receptors in topiramate neuroprotective effects against methylphenidate abuse sequels involving oxidative stress and inflammation in rat isolated hippocampus. Eur. J. Pharmacol. 2016, 784, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, S.X.; Li, J.R.; Du, H.G.; Wang, C.H.; Zhang, J.M.; Wu, Q. Topiramate attenuates early brain injury following subarachnoid haemorrhage in rats via duplex protection against inflammation and neuronal cell death. Brain Res. 2015, 1622, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.M.; Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, M.J.; Choi, H.C.; Song, H.K.; Kang, T.C. Up-regulation of endothelial endothelin-1 expression prior to vasogenic edema formation in the rat piriform cortex following status epilepticus. Neurosci. Lett. 2011, 501, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. Roscovitine attenuates microglia activation and monocyte infiltration via p38 MAPK inhibition in the rat frontoparietal cortex following status epilepticus. Cells 2019, 8, 746. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.A.; McCoy, M.K.; Sporn, M.B.; Tansey, M.G. The synthetic triterpenoid CDDO-methyl ester modulates microglial activities, inhibits TNF production, and provides dopaminergic neuroprotection. J. Neuroinflammation 2008, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Takagi, T.; Kitashoji, A.; Yamauchi, K.; Shimazawa, M.; Hara, H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol. Dis. 2016, 89, 136–146. [Google Scholar] [CrossRef]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-Me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me selectively attenuates CA1 neuronal death induced by status epilepticus via facilitating mitochondrial fission independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Park, H.; Choi, S.H.; Kong, M.J.; Kim, J.E.; Kang, T.C. CDDO-Me attenuates vasogenic edema and astroglial death by regulating NF-κB p65 phosphorylations and Nrf2 expression following status epilepticus. Int. J. Mol. Sci. 2019, 20, 4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Zhang, X.H.; Peng, L.; Liu, Z.; Yang, Y.X.; He, Z.X.; Dang, H.W.; Zhou, S.F. Bardoxolone methyl (CDDO-Me or RTA402) induces cell cycle arrest, apoptosis and autophagy via PI3K/Akt/mTOR and p38 MAPK/Erk1/2 signaling pathways in K562 cells. Am. J. Transl. Res. 2017, 9, 4652–4672. [Google Scholar]
- Deeb, D.; Gao, X.; Jiang, H.; Dulchavsky, S.A.; Gautam, S.C. Oleanane triterpenoid CDDO-Me inhibits growth and induces apoptosis in prostate cancer cells by independently targeting pro-survival Akt and mTOR. Prostate 2009, 69, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Zhe, H.; Zhao, R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yore, M.M.; Liby, K.T.; Honda, T.; Gribble, G.W.; Sporn, M.B. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol. Cancer Ther. 2006, 5, 3232–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Fuchs, R.J.; Malhotra, D.; Scollick, C.; Traore, K.; Bream, J.H.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Kensler, T.W.; et al. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid. Redox Signal. 2007, 9, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Rounds, B.V.; Gribble, G.W.; Suh, N.; Wang, Y.; Sporn, M.B. Design and synthesis of 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8, 2711–2714. [Google Scholar] [CrossRef]
- Eitas, T.K.; Stepp, W.H.; Sjeklocha, L.; Long, C.V.; Riley, C.; Callahan, J.; Sanchez, Y.; Gough, P.; Knowlin, L.; van Duin, D.; et al. Differential regulation of innate immune cytokine production through pharmacological activation of Nuclear Factor-Erythroid-2-Related Factor 2 (NRF2) in burn patient immune cells and monocytes. PLoS ONE 2017, 12, e0184164. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. BEACON Trial Investigators. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/ endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramprasad, M.P.; Terpstra, V.; Kondratenko, N.; Quehenberger, O.; Steinberg, D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 14833–14838. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, J.; Funakoshi-Tago, M.; Tago, K.; Mashino, T.; Inoue, H.; Sonoda, Y.; Kasahara, T. Licochalcone A significantly suppresses LPS signaling pathway through the inhibition of NF-κB p65 phosphorylation at serine 276. Cell Signal 2009, 21, 778–785. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, J.E.; Kim, Y.J.; Kim, M.J.; Kang, T.C. Hyperforin attenuates microglia activation and inhibits p65-Ser276 NFκB phosphorylation in the rat piriform cortex following status epilepticus. Neurosci. Res. 2014, 85, 39–50. [Google Scholar] [CrossRef]
- Picascia, A.; Grimaldi, V.; Iannone, C.; Soricelli, A.; Napoli, C. Innate and adaptive immune response in stroke: Focus on epigenetic regulation. J. Neuroimmunol. 2015, 289, 111–120. [Google Scholar] [CrossRef]
- Abe, N.; Choudhury, M.E.; Watanabe, M.; Kawasaki, S.; Nishihara, T.; Yano, H.; Matsumoto, S.; Kunieda, T.; Kumon, Y.; Yorozuya, T.; et al. Comparison of the detrimental features of microglia and infiltrated macrophages in traumatic brain injury: A study using a hypnotic bromovalerylurea. Glia 2018, 66, 2158–2173. [Google Scholar] [CrossRef]
- Camer, D.; Yu, Y.; Szabo, A.; Fernandez, F.; Dinh, C.H.L.; Huang, X.F. Bardoxolone methyl prevents high-fat diet-induced alterations in prefrontal cortex signaling molecules involved in recognition memory. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2015, 59, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Yuhki, N.; Moore, S.K.; Appella, E.; Lerman, M.I.; Leonard, E.J. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 1989, 244, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Fung, Y.K.; Liu, X.; Pahan, K. Up-regulation of microglial CD11b expression by nitric oxide. J. Biol. Chem. 2006, 281, 14971–14980. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.M.; Zong, Y.; Sun, L.; Guo, J.Z.; Zhang, W.; He, Y.; Song, R.; Wang, W.M.; Xiao, C.J.; Lu, D. Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS ONE 2012, 7, e32195. [Google Scholar] [CrossRef] [Green Version]
- Morganti, J.M.; Goulding, D.S.; Van Eldik, L.J. Deletion of p38α MAPK in microglia blunts trauma-induced inflammatory responses in mice. J Neuroinflammation 2019, 16, 98. [Google Scholar] [CrossRef] [Green Version]
- Katayama, T.; Kobayashi, H.; Okamura, T.; Yamasaki-Katayama, Y.; Kibayashi, T.; Kimura, H.; Ohsawa, K.; Kohsaka, S.; Minami, M. Accumulating microglia phagocytose injuredneurons in hippocampal slice cultures: Involvement of p38 MAP kinase. PLoS ONE 2012, 7, e40813. [Google Scholar] [CrossRef]
- Castrillo, A.; de Las Heras, B.; Hortelano, S.; Rodriguez, B.; Villar, A.; Bosca, L. Inhibition of the nuclear factor kappa B (NF-kappa B) pathway by tetracyclic kaurene diterpenes in macrophages. Specific effects on NF-kappa B-inducing kinase activity and on the coordinate activation of ERK and p38 MAPK. J. Biol. Chem. 2001, 276, 15854–15860. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Richmond, A. Nuclear factor-kappa B activation by the CXC chemokine melanoma growth-stimulatory activity/growth-regulated protein involves the MEKK1/p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 2001, 276, 3650–3659. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ford, H.R.; Grishin, A.V. NF-kappaB-mediated expression of MAPK phosphatase-1 is an early step in desensitization to TLR ligands in enterocytes. Mucosal Immunol. 2010, 3, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Uesugi, M.; Nakajima, K.; Tohyama, Y.; Kohsaka, S.; Kurihara, T. Nonparticipation of nuclear factor kappa B (NFkappaB) in the signaling cascade of c-Jun N-terminal kinase (JNK)- and p38 mitogen-activated protein kinase (p38MAPK)-dependent tumor necrosis factor alpha (TNFalpha) induction in lipopolysaccharide (LPS)-stimulated microglia. Brain Res. 2006, 1073–1074, 48–59. [Google Scholar]
- Škandík, M.; Mrvová, N.; Bezek, Š.; Račková, L. Semisynthetic quercetin-quinone mitigates BV-2 microglia activation through modulation of Nrf2 pathway. Free Radic. Biol. Med. 2020, 152, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Galán-Ganga, M.; Del Río, R.; Jiménez-Moreno, N.; Díaz-Guerra, M.; Lastres-Becker, I. Cannabinoid CB(2) Receptor Modulation by the Transcription Factor NRF2 is Specific in Microglial Cells. Cell. Mol. Neurobiol. 2020, 40, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Ikeda, Y.; Ohta, Y.; Deguchi, K.; Tian, F.; Shang, J.; Matsuura, T.; Abe, K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011, 1370, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Brandenburg, L.O.; Rosen, C.; Fragoulis, A.; Kipp, M.; Pufe, T.; Beyer, C.; Wruck, C.J. Nrf2 expression by neurons, astroglia, and microglia in the cerebral cortical penumbra of ischemic rats. J. Mol. Neurosci. 2012, 46, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Li, J.; Meyer, C.J.; Janicki, J.S.; Hannink, M.; Cui, T. Dihydro-CDDO-trifluoroethyl amide (dh404), a novel Nrf2 activator, suppresses oxidative stress in cardiomyocytes. PLoS ONE 2009, 4, e8391. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Paonessa, J.D.; Zhang, Y. Mechanism of chemical activation of Nrf2. PLoS ONE 2012, 7, e35122. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.; Cha, Y.; Kim, S.; Kim, J. tBHQ inhibits LPS-induced microglial activation via Nrf2-mediated suppression of p38 phosphorylation. Biochem. Biophys. Res. Commun. 2009, 380, 449–453. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| CD68 | Mouse | Abcam (ab31630) | 1:100 (IH) |
| IB4 | Vector (B-1205) | 1:200 (IH) | |
| Iba-1 | Rabbit | Biocare Medical (CP 290) | 1:500 (IH) |
| MCP-1 | Mouse | Abcam (ab25124) | 1:100 (IH) 1:2000 (WB) |
| NFκB | Rabbit | Abcam (ab16502) | 1:1000 (WB) |
| NFκB-S276 | Rabbit | Abcam (ab106129) | 1:100 (IH) |
| Nrf2 | Mouse | Abcam (ab89443) | 1:100 (IH) 1:1000 (WB) |
| p38 MAPK | Rabbit | Cell signaling (#9212) | 1:1000 (WB) |
| phospho-p38 MAPK | Rabbit | Abbiotec (# 251256) | 1:200 (IH) 1:500 (WB) |
| TNF-α | Goat | R&D systems (AF-510-NA) | 1:1000 (IH) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Park, H.; Lee, J.-E.; Kang, T.-C. CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus. Cells 2020, 9, 1123. https://doi.org/10.3390/cells9051123
Kim J-E, Park H, Lee J-E, Kang T-C. CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus. Cells. 2020; 9(5):1123. https://doi.org/10.3390/cells9051123
Chicago/Turabian StyleKim, Ji-Eun, Hana Park, Ji-Eun Lee, and Tae-Cheon Kang. 2020. "CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus" Cells 9, no. 5: 1123. https://doi.org/10.3390/cells9051123
APA StyleKim, J. -E., Park, H., Lee, J. -E., & Kang, T. -C. (2020). CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus. Cells, 9(5), 1123. https://doi.org/10.3390/cells9051123