Intraglomerular Monocyte/Macrophage Infiltration and Macrophage–Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Glomerulus Isolation
2.3. Glycosuria and Proteinuria
2.4. Serum Urea and Creatinine
2.5. Histological Analysis and Immunohistofluorescence
2.6. Transcriptomic Analysis
2.7. Human and Rat Peripheral Blood Mononuclear Cell (PBMCs) Isolation
2.8. Rat Macrophage Migration Assay
2.9. Human Macrophage Transmigration Assay
2.10. Flow Cytometry Assay
2.11. Immunocytofluorescence
2.12. RNA Isolation and RT-qPCR
2.13. Statistical Analysis
3. Results
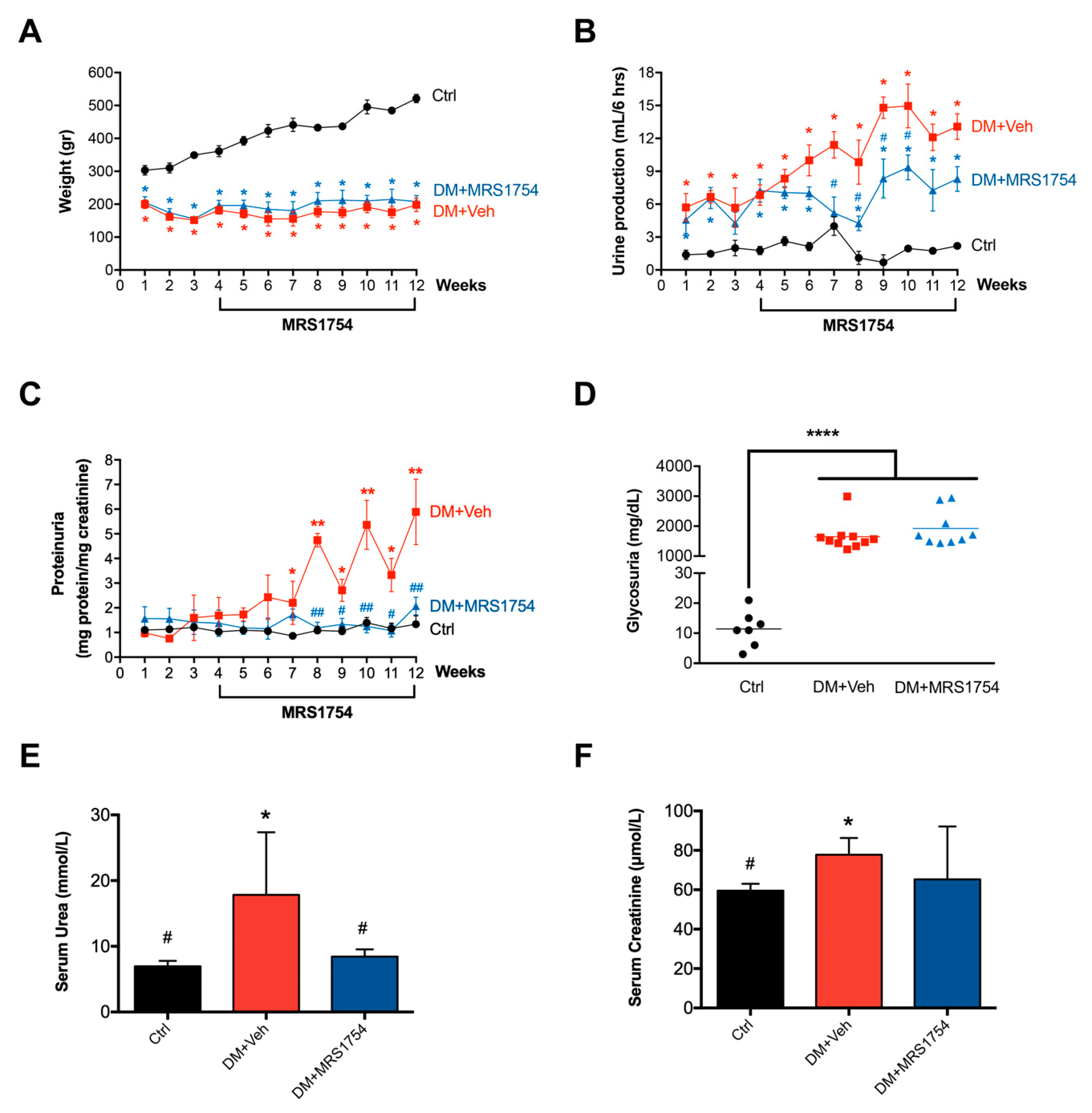
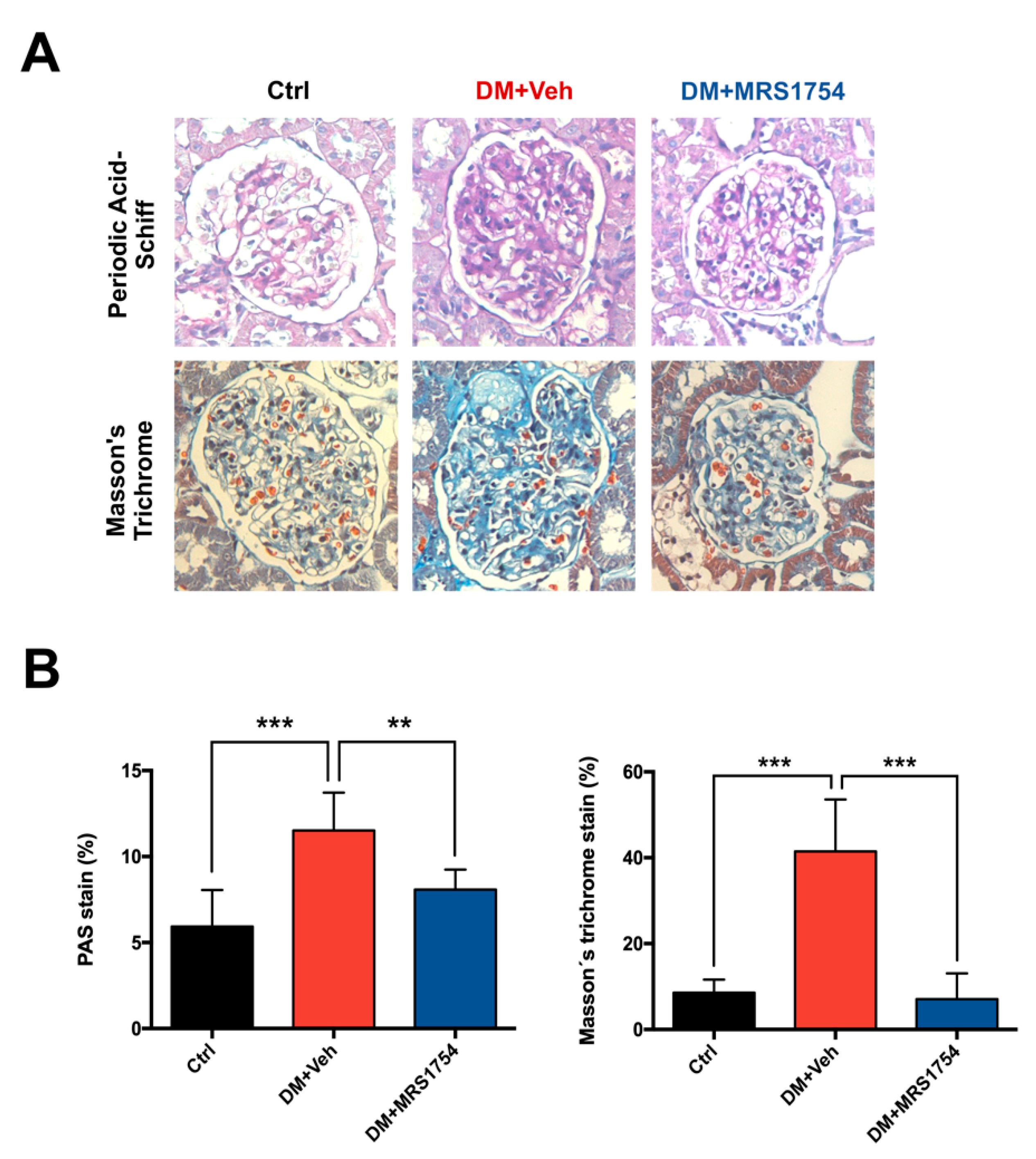
3.1. In Vivo MRS1754 Treatment Attenuates the Clinical Signs of Glomerular Injuries and Decreases Glomerular Fibrosis in Diabetic Rats
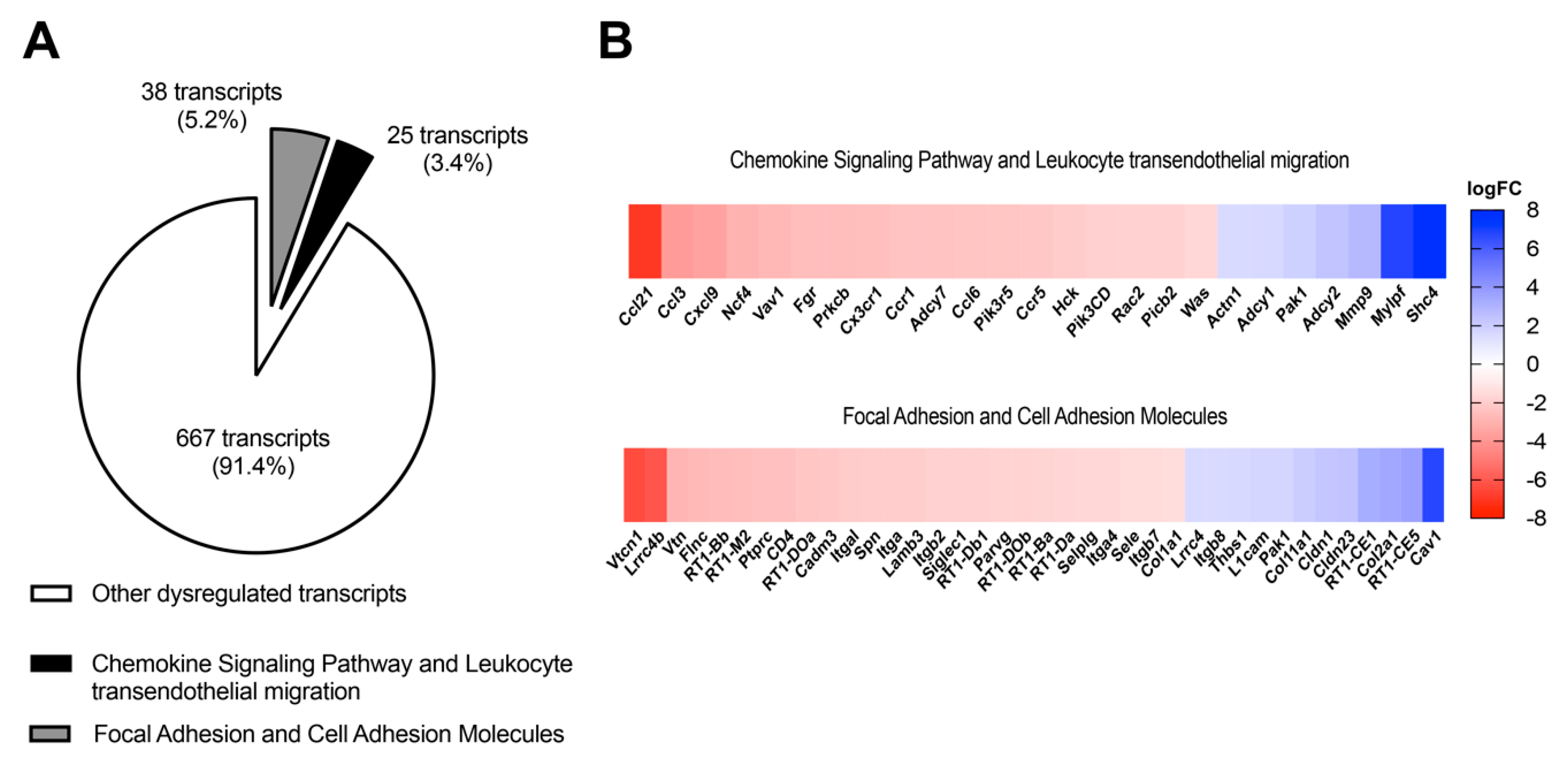
3.2. In Vivo Antagonism of A2BAR in Diabetic Rats Decreases the Transcriptional Expression of Chemokine-Chemoattractant and Cell Adhesion Molecules for Monocytes/Macrophages in Ex Vivo Glomeruli
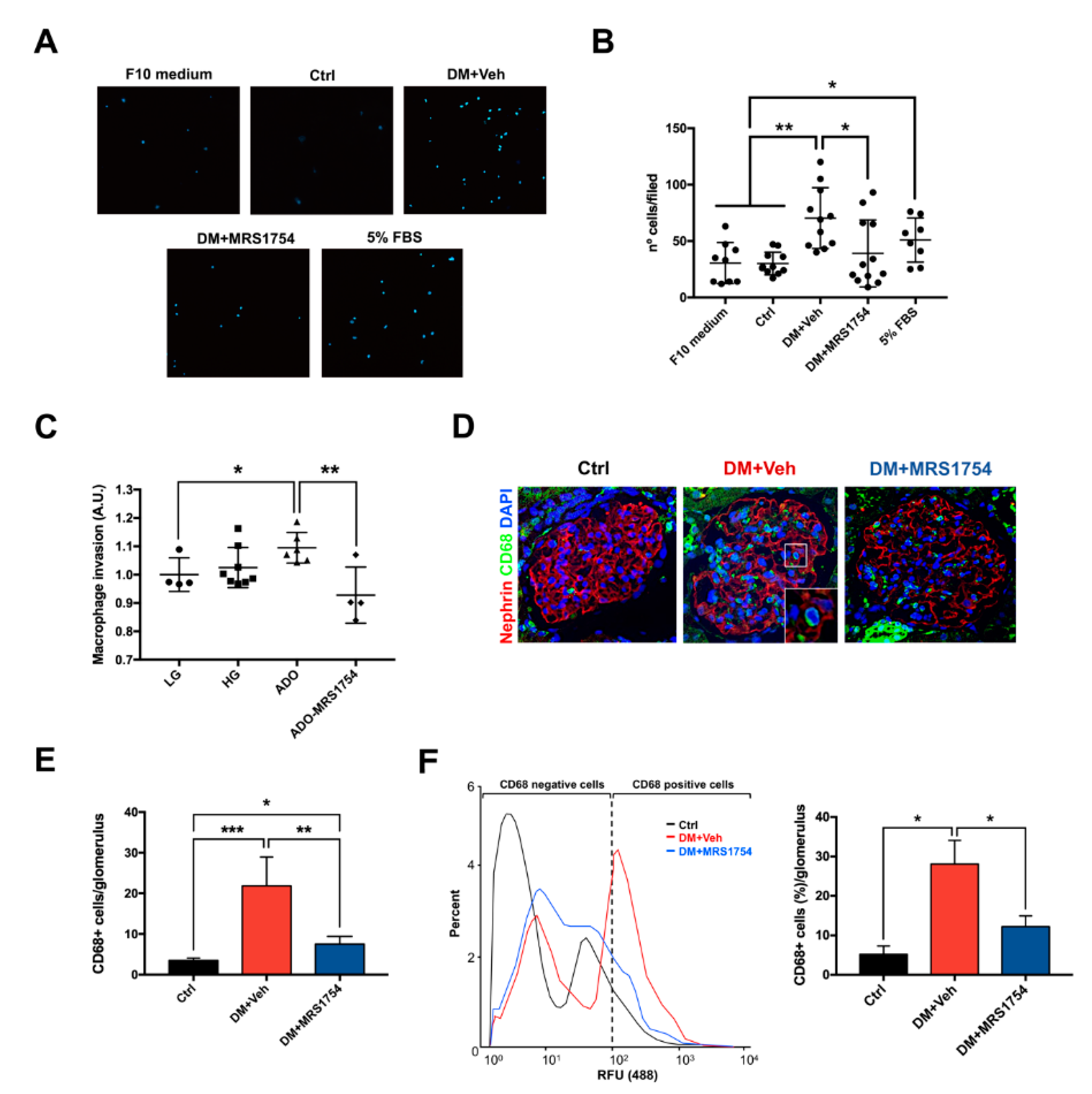
3.3. MRS1754 Treatment Decreases the Migration/Transmigration and Intraglomerular Monocyte/Macrophage Infiltration by Inhibiting the Chemoattractant Effect Induced by Diabetic Rat Glomeruli
3.4. A2BAR Blockade Decreases the Intraglomerular Macrophage–Myofibroblast Transition in Diabetic Rats
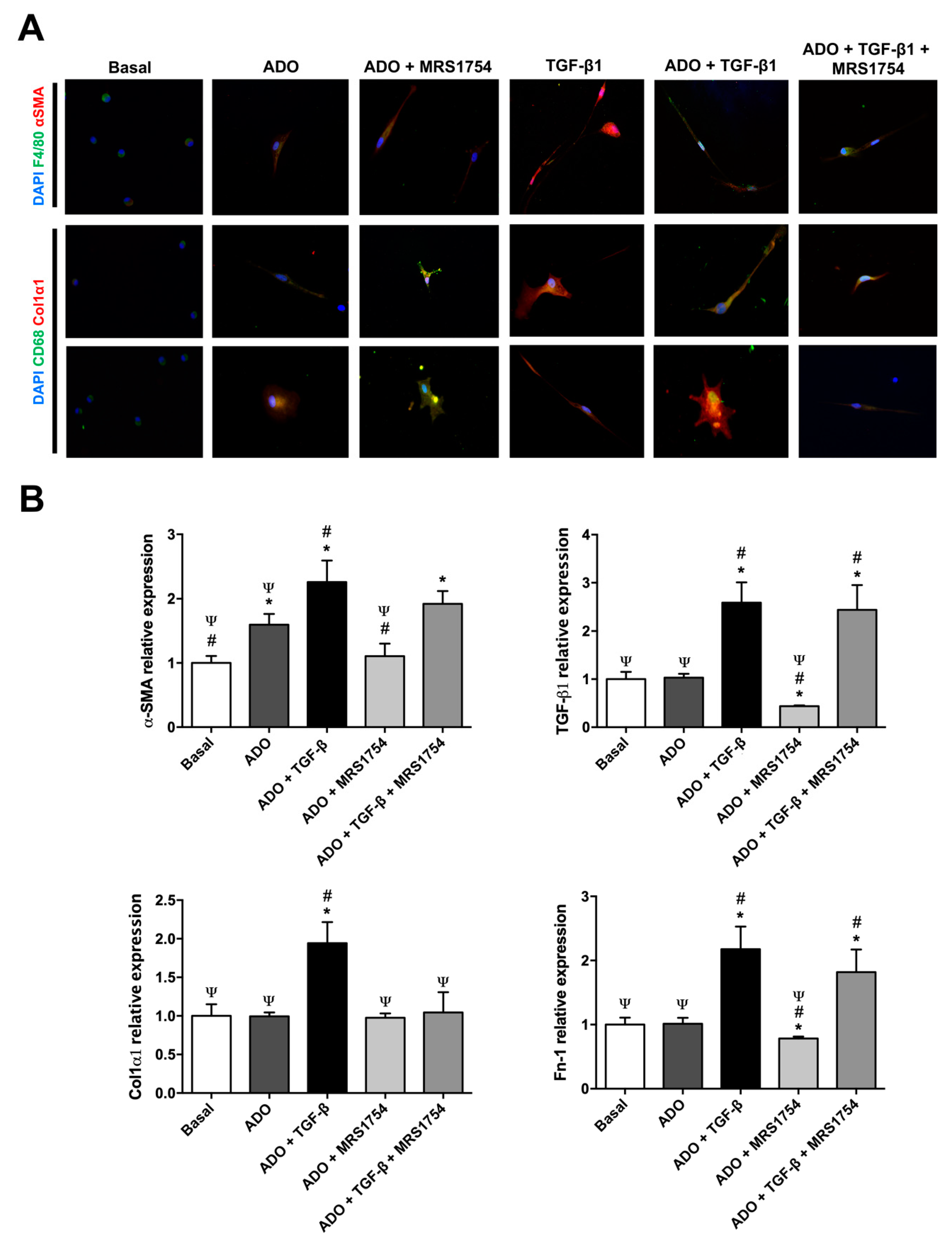
3.5. In Vitro MRS1754 Treatment Reduces Macrophage to Myofibroblast-Like Morphology and MMT Markers in Human Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A. Diabetic nephropathy—Complications and treatment. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global prevalence of chronic kidney disease—A systematic review and meta-analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chaudhari, P.; Yang, H.; Fang, A.; Zhao, J.; Law, E.; Wu, E.; Jiang, R.; Seifeldin, R. Healthcare resource use, costs, and disease progression associated with diabetic nephropathy in adults with type 2 diabetes: A retrospective observational study. Diabetes Therapy 2017, 8, 555–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, I. Cost of treating diabetic kidney disease. Indian J. Nephrol. 2014, 24, 139–140. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Kazancioğlu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.; Bader, H.; Grund, K.E.; Mackensen-Haen, S.; Christ, H.; Bohle, A. Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathol. Res. Pract. 1980, 167, 204–216. [Google Scholar] [CrossRef]
- Dabla, P.K. Renal function in diabetic nephropathy. World J. Diabetes 2010, 1, 48–56. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitson, T.D. Fibrosis in the kidney: Is a problem shared a problem halved. Fibrogenesis Tissue Repair 2012, 5, S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Annemans, L.; Brown, E.; Gansevoort, R.; Gout-Zwart, J.J.; Lameire, N.; Morton, R.L.; Oberbauer, R.; Postma, M.J.; Tonelli, M.; et al. Reducing the costs of chronic kidney disease while delivering quality health care: A call to action. Nat. Rev. Nephrol. 2017, 13, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, L.; Alvarez, P.J.; Reaven, N.L.; Funk, S.E.; McGaughey, K.J.; Romero, A.; Brenner, M.S.; Onuigbo, M. All-cause costs increase exponentially with increased chronic kidney disease stage. Am. J. Manag. Care 2017, 23, S163–S172. [Google Scholar]
- Gheith, O.; Farouk, N.; Nampoory, N.; Halim, M.A.; Al-Otaibi, T. Diabetic kidney disease: World wide difference of prevalence and risk factors. J. Nephropharmacol. 2016, 5, 49–56. [Google Scholar] [CrossRef]
- Vega, G.; Alarcón, S.; San Martín, R. The cellular and signalling alterations conducted by TGF-β contributing to renal fibrosis. Cytokine 2016, 88, 115–125. [Google Scholar] [CrossRef]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.F.; Liu, Y. New insights into the pathogenesis and therapeutics of kidney fibrosis. Kidney Int. Suppl. 2014, 4, 1. [Google Scholar] [CrossRef]
- Yang, H.; Fogo, A.B. Fibrosis and renal aging. Kidney Int. Suppl. 2014, 4, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Nikolic-Paterson, D.; Lan, H. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Han, G.; Li, F.; Singh, T.P.; Wolf, P.; Wang, X.J. The pro-inflammatory role of TGFβ1: A paradox. Int. J. Biol. Sci. 2012, 8, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilling, D.; Vakil, V.; Cox, N.; Gomer, R.H. TNF-α-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 11929–11934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Kim, B.S.; Lee, S.K.; Park, J.S.; Kim, S.B. Interleukin-1beta stimulates the production of extracellular matrix in cultured human peritoneal mesothelial cells. Perit. Dial. Int. 1999, 19, 211–220. [Google Scholar] [CrossRef] [PubMed]
- MacKay, K.; Striker, L.J.; Stauffer, J.W.; Doi, T.; Agodoa, L.Y.; Striker, G.E. Transforming growth factor-beta. Murine glomerular receptors and responses of isolated glomerular cells. J. Clin. Investig. 1989, 83, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Grande, M.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.F.; Chiang, W.C.; Lai, C.F.; Chang, F.C.; Chen, Y.T.; Chou, Y.H.; Wu, T.H.; Linn, G.R.; Ling, H.; Wu, K.D.; et al. Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.W.; Chand, S.; Savage, C.O. Biology of the renal pericyte. Nephrol. Dial. Transpl. 2012, 27, 2149–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Harris, D.C. Macrophages in renal disease. J. Am. Soc. Nephrol. 2011, 22, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Wang, S.; Huang, X.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, X.; Ng, Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.; Xiao, J.; Wang, Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrowderived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Navarro, A.; Guerrero-Hue, M.; Martín-Fernandez, B.; Cortegano, I.; Olivares-Alvaro, E.; de las Heras, N.; Alía, M.; de Andrés, B.; Gaspar, M.L.; Egido, J.; et al. Phenotypic characterization of macrophages from rat kidney by flow cytometry. J. Vis. Exp. 2016, 116, e54599. [Google Scholar] [CrossRef] [PubMed]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2011, 4, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Jiang, H.; Pan, J.; Huang, X.R.; Wang, Y.C.; Huang, H.F.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y.; Chen, J.H. Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [Green Version]
- Lovisa, S.; Zeisberg, M.; Kalluri, R. Partial Epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol. Metab. 2016, 27, 681–695. [Google Scholar] [CrossRef]
- Tan, T.K.; Zheng, G.; Hsu, T.T.; Lee, S.R.; Zhang, J.; Zhao, Y.; Tian, X.; Wang, Y.; Wang, Y.M.; Cao, Q.; et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab. Investig. 2013, 93, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Mariasegaram, M.; Tesch, G.H.; Verhardt, S.; Hurst, L.; Lan, H.Y.; Nikolic-Paterson, D.J. Lefty antagonises TGF-beta1 induced epithelial-mesenchymal transition in tubular epithelial cells. Biochem. Biophys. Res. Commun. 2010, 393, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, J.; Yan, J.; Zhang, L.; Chen, G.; He, L.; Wang, Y. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PLoS ONE 2012, 7, e52415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyarzún, C.; Garrido, W.; Alarcón, S.; Yáñez, A.; Sobrevia, L.; Quezada, C.; San Martín, R. Adenosine contribution to normal renal physiology and chronic kidney disease. Mol. Asp. Med. 2017, 55, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cárdenas, A.; Toledo, C.; Oyarzún, C.; Sepúlveda, A.; Quezada, C.; Guillén-Gómez, E.; Díaz-Encarnación, M.M.; Pastor-Anglada, M.; San Martín, R. Adenosine A(2B) receptor-mediated VEGF induction promotes diabetic glomerulopathy. Lab. Investig. 2013, 93, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.F.; Liang, Q.L.; Liang, X.P.; Wang, Y.M.; Hu, P.; Li, P.; Luo, G.A. Ultraviolet and tandem mass spectrometry for simultaneous quantification of 21 pivotal metabolites in plasma from patients with diabetic nephropathy. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1930–1936. [Google Scholar] [CrossRef]
- Fredholm, B.B. Adenosine—A physiological or pathophysiological agent. J. Mol. Med. 2014, 92, 201–206. [Google Scholar] [CrossRef]
- Perez-Aso, M.; Feig, J.L.; Mediero, A.; Cronstein, B.N. Adenosine A2A receptor and TNF-α regulate the circadian machinery of the human monocytic THP-1 cells. Inflammation 2013, 36, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Roa, H.; Gajardo, C.; Troncoso, E.; Fuentealba, V.; Escudero, C.; Yáñez, A.; Sobrevia, L.; Pastor-Anglada, M.; Quezada, C.; San Martin, R. Adenosine mediates transforming growth factor-beta 1 release in kidney glomeruli of diabetic rats. FEBS Lett. 2009, 583, 3192–3198. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.F.; Farrell, F.X.; Morel, D.; Law, W.; Murphy, S. Adenosine signaling increases proinflammatory and profibrotic mediators through activation of a functional adenosine 2B receptor in renal fibroblasts. Ann. Clin. Lab. Sci. 2016, 46, 339–345. [Google Scholar]
- Patel, L.; Thaker, A. The effects of adenosine A2B receptor inhibition on VEGF and nitric oxide axis-mediated renal function in diabetic nephropathy. Ren. Fail. 2014, 36, 916–924. [Google Scholar] [CrossRef]
- Xie, T.; Li, G.; Yang, H.; Wang, X.; Li, J.; Zhang, W. Abstract 17075: Inhibition of adenosine A2B receptor ameliorates renal fibrosis by regulating macrophage infiltration and polarization. Circulation 2017, 136, A17075. [Google Scholar]
- Alarcón, S.; Garrido, W.; Cappelli, C.; Suárez, R.; Oyarzún, C.; Quezada, C.; San Martín, R. Deficient insulin-mediated Upregulation of the Equilibrative nucleoside transporter 2 contributes to chronically increased adenosine in diabetic Glomerulopathy. Sci. Rep. 2017, 7, 9439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel Wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.; Pramod, A.B.; Croft, M.; Ravichandran, K.S.; Ting, J.P. How mouse macrophages sense what is going on. Front. Immunol. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Tesch, G.H.; Allen, T.J. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology 2007, 12, 261–266. [Google Scholar] [CrossRef]
- Faulhaber-Walter, R.; Chen, L.; Oppermann, M.; Kim, S.M.; Huang, Y.; Hiramatsu, N.; Mizel, D.; Kajiyama, H.; Zerfas, P.; Briggs, J.P.; et al. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J. Am. Soc. Nephrol. 2008, 19, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, M.; Pao, A.C. Adenosine activates a2b receptors and enhances chloride secretion in kidney inner medullary collecting duct cells. Hypertension 2010, 55, 1123–1128. [Google Scholar] [CrossRef]
- Risdon, R.A.; Sloper, J.C.; De Wardener, H.E. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet 1968, 292, 363–366. [Google Scholar] [CrossRef]
- Su, Z.; Widomski, D.; Ma, J.; Namovic, M.; Nikkel, A.; Leys, L.; Olson, L.; Salte, K.; Donnelly-Roberts, D.; Esbenshade, T.; et al. Longitudinal changes in measured glomerular filtration rate, renal fibrosis and biomarkers in a rat model of Type 2 diabetic nephropathy. Am. J. Nephrol. 2016, 44, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Voziyan, P.A.; Hudson, B.G.; Zent, R. Regulation of matrix synthesis, remodeling and accumulation in Glomerulosclerosis. Curr. Pharm. Des. 2009, 15, 1318–1333. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, W.; Wen, J.; Zhang, Y.; Kellems, R.E.; Xia, Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD. J. Am. Soc. Nephrol. 2011, 22, 890–901. [Google Scholar] [CrossRef] [Green Version]
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. Macrophage phenotype in kidney injury and repair. Kidney Dis. 2015, 1, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Mak, T.S.; Lan, H.Y. Macrophages in renal fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 285–303. [Google Scholar] [CrossRef]
- Chen, T.; Cao, Q.; Wang, Y.; Harris, D. M2 macrophages in kidney disease: Biology, therapies, and perspectives. Kidney Int. 2019, 95, 760–773. [Google Scholar] [CrossRef]
- Hattori, M.; Nikolic-Paterson, D.J.; Miyazaki, K.; Isbel, N.M.; Lan, H.Y.; Atkins, R.C.; Kawaguchi, H.; Ito, K. Mechanisms of glomerular macrophage infiltration in lipid-induced renal injury. Kidney Int. Suppl. 1999, 71, S47–S50. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T.; Yamabe, H.; Shimada, M.; Murakami, R.; Kumasaka, R.; Nakamura, N.; Osawa, H.; Okumura, K. Thrombin enhances the production of monocyte chemoattractant protein-1 and macrophage inflammatory protein-2 in cultured rat glomerular epithelial cells. Nephrol. Dial. Transpl. 2008, 23, 3412–3417. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Doe, N.; Tan, L.C. Monocyte chemoattractant protein-1 levels in patients with glomerular disease. Am. J. Kidney Dis. 1996, 27, 640–646. [Google Scholar] [CrossRef]
- Nikolic-Paterson, D.J.; Atkins, R.C. The role of macrophages in glomerulonephritis. Nephrol. Dial. Transpl. 2001, 16, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, S.J.; Lee, E.S.; Kim, G.T.; Hyun, M.; Lee, J.H.; Chen, S.; Choi, R.; Kim, H.M.; Lee, E.Y.; Chung, C.H. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol. Dial. Transpl. 2013, 28, 1700–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesch, G.H. MCP-1/CCL2: A new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2008, 294, F697–F701. [Google Scholar] [CrossRef]
- Wendland, M.; Willenzon, S.; Kocks, J.; Davalos-Misslitz, A.C.; Hammerschmidt, S.I.; Schumann, K.; Kremmer, E.; Sixt, M.; Hoffmeyer, A.; Pabst, O.; et al. Lymph node T cell homeostasis relies on steady state homing of dendritic cells. Immunity 2011, 35, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Sakai, N.; Matsushima, K.; Kaneko, S. Fibrocytes: A new insight into kidney fibrosis. Kidney Int. 2007, 72, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, K.; Wada, T.; Furuichi, K.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Takeya, M.; Kuziel, W.A.; Matsushima, K.; Mukaida, N.; et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am. J. Pathol. 2004, 165, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, Z.; Jia, L.; Wang, Y. Role of bone marrow-derived fibroblasts in renal fibrosis. Front. Physiol. 2016, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Wada, T.; Yokoyama, H.; Lipp, M.; Ueha, S.; Matsushima, K.; Kaneko, S. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14098–14103. [Google Scholar] [CrossRef] [Green Version]
- Banas, B.; Wörnle, M.; Berger, T.; Nelson, P.J.; Cohen, C.D.; Kretzler, M.; Pfirstinger, J.; Mack, M.; Lipp, M.; Gröne, H.J.; et al. Roles of SLC/CCL21 and CCR7 in human kidney for mesangial proliferation, migration, apoptosis, and tissue homeostasis. J. Immunol. 2002, 168, 4301–4307. [Google Scholar] [CrossRef] [Green Version]
- Banas, B.; Wörnle, M.; Merkle, M.; Gonzalez-Rubio, M.; Schmid, H.; Kretzler, M.; Pietrzyk, M.C.; Fink, M.; Perez de Lema, G.; Schlöndorff, D. Binding of the chemokine SLC/CCL21 to its receptor CCR7 increases adhesive properties of human mesangial cells. Kidney Int. 2004, 66, 2256–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiño-Rivas, L.; Gonzalez-Lafuente, L.; Sanz, A.; Ruiz-Ortega, M.; Ortiz, A.; Sanchez-Niño, M.D. Non-canonical NFκB activation promotes chemokine expression in podocytes. Sci. Rep. 2016, 6, 28857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse origins of the myofibroblast—Implications for kidney fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Fan, J.M.; Mu, W.; Nikolic-Paterson, D.J.; Yang, W.C.; Huang, T.P.; Atkins, R.C.; Lan, H.Y. Glomerular epithelial-myofibroblast transdifferentiation in the evolution of glomerular crescent formation. Nephrol. Dial. Transpl. 1999, 14, 2860–2872. [Google Scholar] [CrossRef] [Green Version]
- Fujigaki, Y.; Sun, D.F.; Fujimoto, T.; Suzuki, T.; Goto, T.; Yonemura, K.; Morioka, T.; Yaoita, E.; Hishida, A. Mechanisms and kinetics of Bowman’s epithelial-myofibroblast transdifferentiation in the formation of glomerular crescents. Nephron 2002, 92, 203–212. [Google Scholar] [CrossRef]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.M.; Ng, Y.Y.; Hill, P.A.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999, 56, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Ninomiya, K.; Takahashi, A.; Fujioka, Y.; Ishikawa, Y.; Yokoyama, M. Transforming growth factor-beta signaling enhances transdifferentiation of macrophages into smooth muscle-like cells. Hypertens. Res. 2006, 29, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Stachelek, S.J.; Tomczyk, N.; Finley, M.J.; Composto, R.J.; Eckmann, D.M. Correlating macrophage morphology and cytokine production resulting from biomaterial contact. J. Biomed. Mater. Res. 2013, 101, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expet Rev. Mol. Med. 2011, 13, e27. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M.; Costell, M.; Alman, B.A.; Genot, E.; Hinz, B. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J. Cell Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef]
- Kollmannsberger, P.; Bidan, C.M.; Dunlop, J.; Fratzl, P.; Vogel, V. Tensile forces drive a reversible fibroblast-to-myofibroblast transition during tissue growth in engineered clefts. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Bing, Q.; Li, S.; Han, B.; Lu, J.; Baiyun, R.; Zhang, X.; Lv, Y.; Wu, H.; Zhang, Z. Role of A2B adenosine receptor-dependent adenosine signaling in multi-walled carbon nanotube-triggered lung fibrosis in mice. J. Nanobiotechnol. 2019, 17, 45. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, Á.; Muñoz, K.; Nahuelpán, Y.; R. Saez, A.-P.; Mendoza, P.; Jara, C.; Cappelli, C.; Suarez, R.; Oyarzún, C.; Quezada, C.; et al. Intraglomerular Monocyte/Macrophage Infiltration and Macrophage–Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor. Cells 2020, 9, 1051. https://doi.org/10.3390/cells9041051
Torres Á, Muñoz K, Nahuelpán Y, R. Saez A-P, Mendoza P, Jara C, Cappelli C, Suarez R, Oyarzún C, Quezada C, et al. Intraglomerular Monocyte/Macrophage Infiltration and Macrophage–Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor. Cells. 2020; 9(4):1051. https://doi.org/10.3390/cells9041051
Chicago/Turabian StyleTorres, Ángelo, Katherin Muñoz, Yessica Nahuelpán, Angelo-Paolo R. Saez, Pablo Mendoza, Claudia Jara, Claudio Cappelli, Raibel Suarez, Carlos Oyarzún, Claudia Quezada, and et al. 2020. "Intraglomerular Monocyte/Macrophage Infiltration and Macrophage–Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor" Cells 9, no. 4: 1051. https://doi.org/10.3390/cells9041051
APA StyleTorres, Á., Muñoz, K., Nahuelpán, Y., R. Saez, A. -P., Mendoza, P., Jara, C., Cappelli, C., Suarez, R., Oyarzún, C., Quezada, C., & San Martín, R. (2020). Intraglomerular Monocyte/Macrophage Infiltration and Macrophage–Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor. Cells, 9(4), 1051. https://doi.org/10.3390/cells9041051