Why Should Growth Hormone (GH) Be Considered a Promising Therapeutic Agent for Arteriogenesis? Insights from the GHAS Trial




Abstract
:
1. Introduction
2. Vascular Homeostasis: Role of the GH/IGFI Axis
3. Molecular Roles of GH/IGF-I in the Vascular Wall that Favor Arteriogenesis
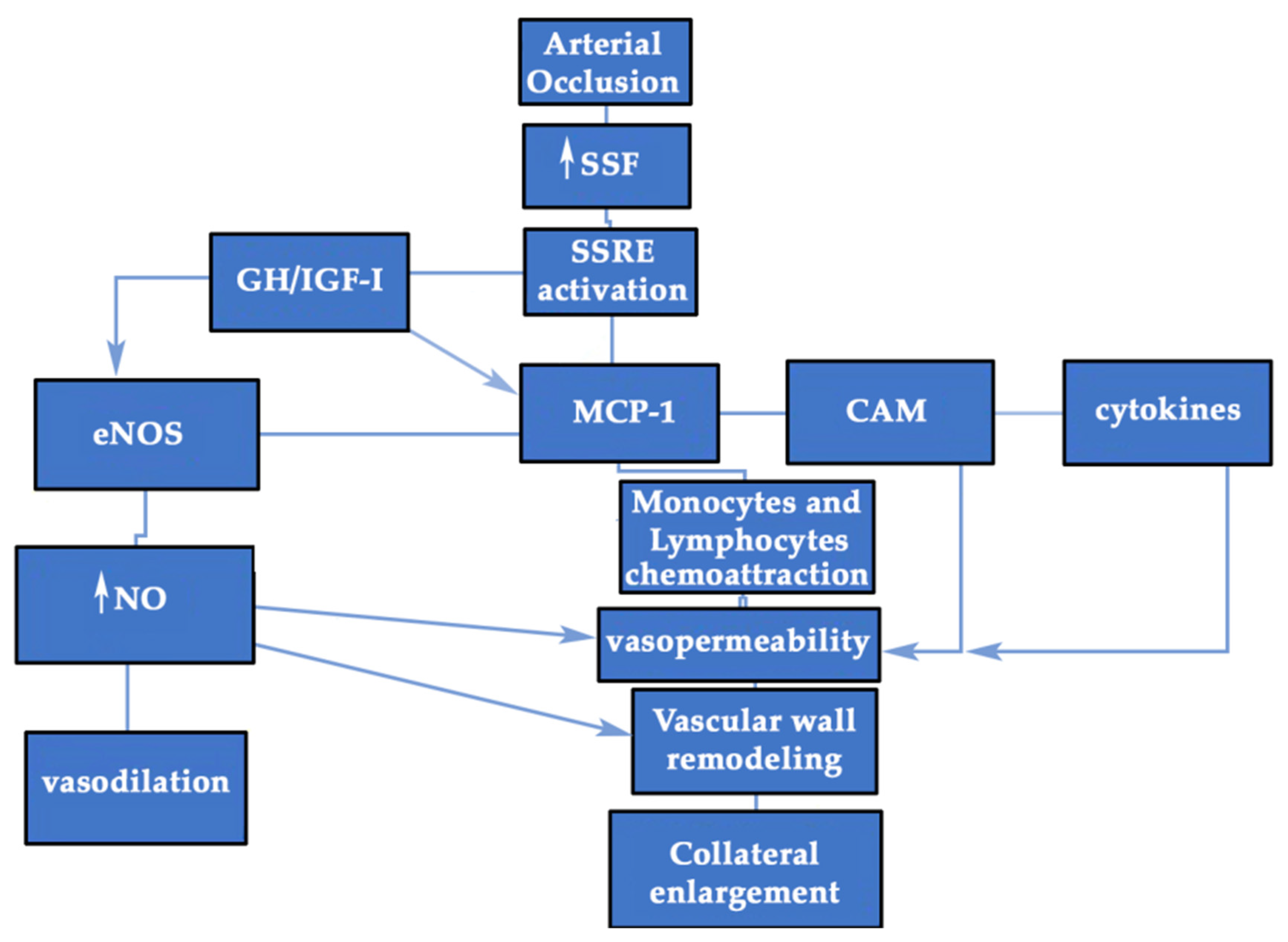
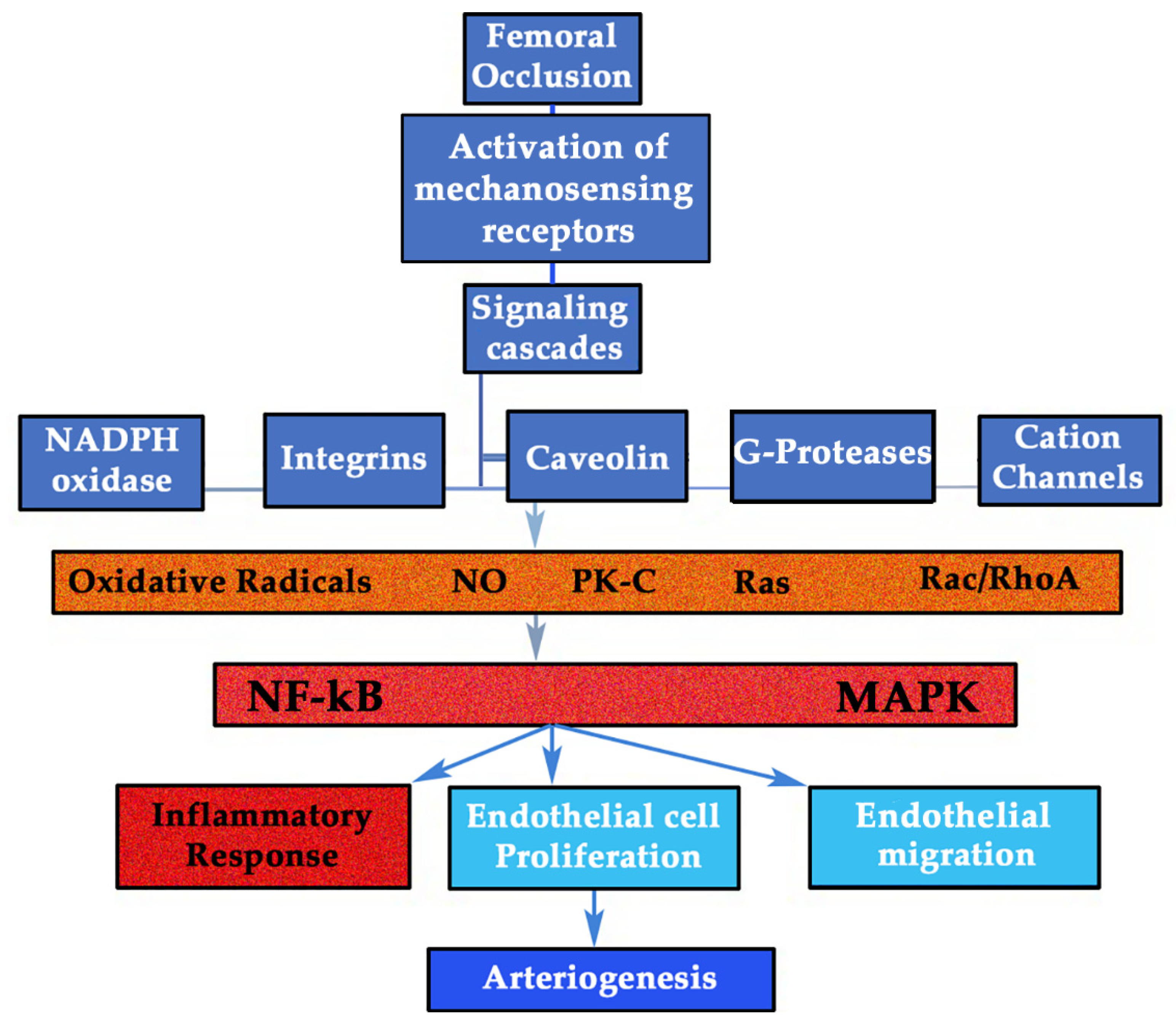
3.1. GH/IGF-I Response to Shear Stress Forces (SSF): the Mechanosensing Pathway
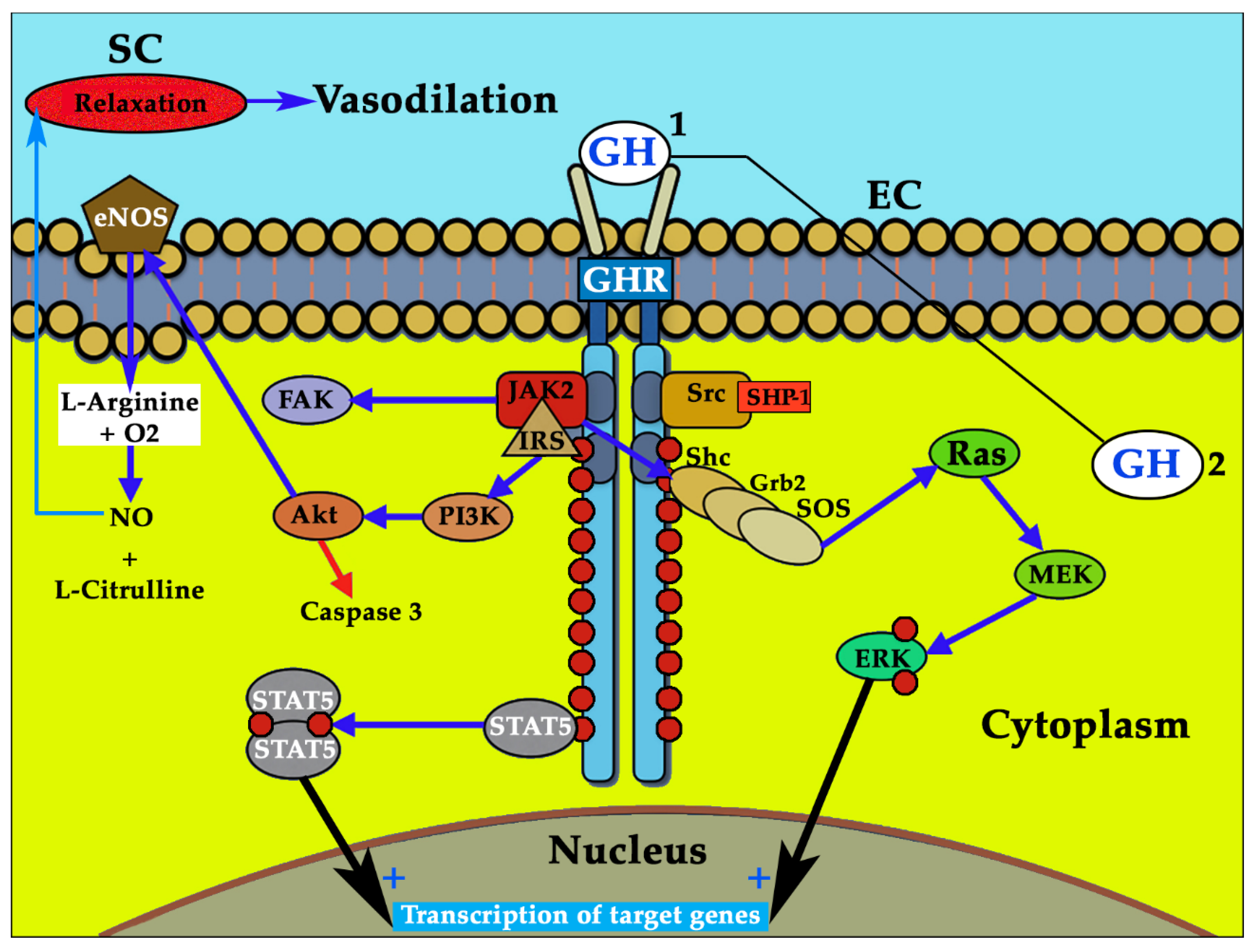
3.2. GH and GHR: a Complex Regulation that Explains Different Results in the Studies Performed
3.3. GH/IGF-I may Favor Inflammation during Collateral Enlargement
3.4. GH/IGF-I and eNOS in Arteriogenesis. Insights from the GHAS Trial: is Redox Balance the Main Actor?
3.5. GH and CXCL12 (SDF-1): A Potential Collaboration for Collateral Growth
3.6. GH and Mesenchymal Stem Cells (MSCs)
3.7. GH and Extracellular Matrix (ECM)
3.8. GH and NO-Independent Vascular Tone: the Role of Sympathetic System
3.9. GH and Midkine (MDK) Relation should Be Investigated
3.10. GH and Klotho: the Perfect Combination?
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Devesa, J.; Almengló, C.; Devesa, P. Multiple Effects of Growth Hormone in the Body: Is It Really the Hormone for Growth? Clin. Med. Insights Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, M.; Verhovec, R.; Zizek, B.; Prezelj, J.; Poredoz, P.; Clayton, R.N. Growth Hormone (GH) Treatment Reverses Early Atherosclerotic Changes in GH-Deficient Adults. J. Clin. Endocrinol. Metab. 1999, 84, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yanagi, K.; Shimizu, M.; Wakamatsu, S.; Niitani, T.; Hosonuma, S.; Sagara, M.; Aso, Y. Effect of Growth Hormone Replacement Therapy on Plasma Diacron-Reactive Oxygen Metabolites and Endothelial Function in Japanese Patients: The GREAT Clinical Study. Endocr. J. 2017, 65, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosén, T.; Bengtsson, B.A. Premature Mortality Due to Cardiovascular Disease in Hypopituitarism. Lancet (London, England) 1990, 336, 285–288. [Google Scholar] [CrossRef]
- Smith, J.C.; Evans, L.M.; Wilkinson, I.; Goodfellow, J.; Cockcroft, J.R.; Scanlon, M.F.; Davies, J.S. Effects of GH Replacement on Endothelial Function and Large-Artery Stiffness in GH-Deficient Adults: A Randomized, Double-Blind, Placebo-Controlled Study. Clin. Endocrinol. (Oxf.) 2002, 56, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Strazhesko, I.D.; Tkacheva, O.N.; Akasheva, D.U.; Dudinskaya, E.N.; Plokhova, E.V.; Pykhtina, V.S.; Kruglikova, A.S.; Brailova, N.V.; Sharashkina, N.V.; Kashtanova, D.A.; et al. Growth Hormone, Insulin-Like Growth Factor-1, Insulin Resistance, and Leukocyte Telomere Length as Determinants of Arterial Aging in Subjects Free of Cardiovascular Diseases. Front. Genet. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Del Rincon, J.P.; Jahn, L.A.; Wu, Y.; Gaylinn, B.; Thorner, M.O.; Liu, Z. Growth Hormone Exerts Acute Vascular Effects Independent of Systemic or Muscle Insulin-like Growth Factor I. J. Clin. Endocrinol. Metab. 2008, 93, 1379–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thum, T.; Tsikas, D.; Frölich, J.C.; Borlak, J. Growth Hormone Induces ENOS Expression and Nitric Oxide Release in a Cultured Human Endothelial Cell Line. FEBS Lett. 2003, 555, 567–571. [Google Scholar] [CrossRef]
- Colao, A. The GH-IGF-I Axis and the Cardiovascular System: Clinical Implications. Clin. Endocrinol. 2008, 347–358. [Google Scholar] [CrossRef]
- Corbacho, A.M.; Martínez de la Escalera, G.; Clapp, C. Roles of Prolactin and Related Members of the Prolactin/Growth Hormone/Placental Lactogen Family in Angiogenesis. J. Endocrinol. 2002, 219–238. [Google Scholar] [CrossRef] [Green Version]
- Brunet-Dunand, S.E.; Vouyovitch, C.; Araneda, S.; Pandey, V.; Vidal, L.J.P.; Print, C.; Mertani, H.C.; Lobie, P.E.; Perry, J.K. Autocrine Human Growth Hormone Promotes Tumor Angiogenesis in Mammary Carcinoma. Endocrinology 2009, 150, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Abs, R. Long-Term Growth Hormone Replacement Therapy in Hypopituitary Adults. Drugs 2002, 62, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.M.; Davies, J.S.; Anderson, R.A.; Ellis, G.R.; Jackson, S.K.; Lewis, M.J.; Frenneaux, M.P.; Rees, A.; Scanlon, M.F. The Effect of GH Replacement Therapy on Endothelial Function and Oxidative Stress in Adult Growth Hormone Deficiency. Eur. J. Endocrinol. 2000, 142, 254–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, R.; Guardasole, V.; Angelini, V.; D’Amico, F.; Zarra, E.; Matarazzo, M.; Sacca, L. Acute Effects of Growth Hormone on Vascular Function in Human Subjects. J. Clin. Endocrinol. Metab. 2003, 88, 2817–2820. [Google Scholar] [CrossRef]
- Elhadd, T.A.; Abdu, T.A.; Oxtoby, J.; Kennedy, G.; McLaren, M.; Neary, R.; Belch, J.J.F.; Clayton, R.N. Biochemical and Biophysical Markers of Endothelial Dysfunction in Adults with Hypopituitarism and Severe GH Deficiency. J. Clin. Endocrinol. Metab. 2001, 86, 4223–4232. [Google Scholar] [CrossRef]
- Caicedo, D.; Díaz, O.; Devesa, P.; Devesa, J. Growth Hormone (GH) and Cardiovascular System. Int. J. Mol. Sci. 2018, 19, 290. [Google Scholar] [CrossRef] [Green Version]
- Palmeiro, C.R.; Anand, R.; Dardi, I.K.; Balasubramaniyam, N.; Schwarcz, M.D.; Weiss, I.A. Growth Hormone and the Cardiovascular System. Cardiol. Rev. 2012, 20, 197–207. [Google Scholar] [CrossRef]
- Wahlberg, E. Angiogenesis and Arteriogenesis in Limb Ischemia. J. Vasc. Surg. 2003, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Veldhuis, J.D.; Roemmich, J.N.; Richmond, E.J.; Bowers, C.Y. Somatotropic and Gonadotropic Axes Linkages in Infancy, Childhood, and the Puberty-Adult Transition. Endocr. Rev. 2006, 27, 101–140. [Google Scholar] [CrossRef] [Green Version]
- Arce, V.M.; Devesa, J.; Fernández-Tresguerres, J.A. Hormona de Crecimiento. In Fisiología Humana; Fernández-Tresguerres, J.A., Ed.; Mcgraw-Hill Medical: Madrid, Spain, 2019; pp. 1–36. [Google Scholar]
- Drake, C.J. Embryonic and Adult Vasculogenesis. Birth Defects Res. Part C-Embryo Today Rev 2003, 73–82. [Google Scholar] [CrossRef]
- Groothuis, P.G. Angiogenesis and Vascular Remodelling in Female Reproductive Organs. Angiogenesis 2005, 8, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of Angiogenesis by Hypoxia: Role of the HIF System. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Skamira, C.; Bode-Böger, S.M.; Brabant, G.; Von Zur Mühlen, A.; Frölich, J.C. Nitric Oxide May Mediate the Hemodynamic Effects of Recombinant Growth Hormone in Patients with Acquired Growth Hormone Deficiency: A Double-Blind, Placebo-Controlled Study. J. Clin. Investig. 1996, 98, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, H.; Gordienko, D.V.; Tonshoff, B.; Gelato, M.C.; Goligorsky, M.S. Direct Demonstration of Insulin-Like Growth Factor-I-Induced Nitric Oxide Production by Endothelial Cells. Kidney Int. 1994, 45, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, R.S.; Boes, M.; Dake, B.L.; Booth, B.A.; Henley, S.A.; Sandra, A. Insulin, Insulin-Like Growth Factors, and Vascular Endothelium. Am. J. Med. 1988, 85, 59–70. [Google Scholar] [CrossRef]
- Wickman, A.; Jonsdottir, I.; Bergstrom, G.; Hedin, L. GH and IGF-I Regulate the Expression of Endothelial Nitric Oxide Synthase (ENOS) in Cardiovascular Tissues of Hypophysectomized Female Rats. Eur. J. Endocrinol. 2002, 147, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Warzecha, Z.; Dembiński, A.; Ceranowicz, P.; Konturek, S.J.; Tomaszewska, R.; Stachura, J.; Konturek, P.C. IGF-1 Stimulates Production of Interleukin-10 and Inhibits Development of Caerulein-Induced Pancreatitis. J. Physiol. Pharmacol. 2003, 54, 575–590. [Google Scholar]
- Shimizu, Y.; Nagaya, N.; Teranishi, Y.; Imazu, M.; Yamamoto, H.; Shokawa, T.; Kangawa, K.; Kohno, N.; Yoshizumi, M. Ghrelin Improves Endothelial Dysfunction through Growth Hormone-Independent Mechanisms in Rats. Biochem. Biophys. Res. Commun. 2003, 310, 830–835. [Google Scholar] [CrossRef]
- Waters, M.J. The Growth Hormone Receptor. Growth Horm. IGF Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. (Lausanne) 2018, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Bauersachs, J. Growth Hormone Regulates Vascular Function—What We Know from Bench and Bedside. Eur. J. Clin. Pharmacol. 2006. [Google Scholar] [CrossRef]
- Tanimura, S.; Takeda, K. ERK Signalling as a Regulator of Cell Motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, T.; Goh, E.L.K.; Lobie, P.E. Growth Hormone Stimulates the Tyrosine Phosphorylation and Association of P125 Focal Adhesion Kinase (FAK) with JAK2. J. Biol. Chem. 1998, 273, 10682–10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.; Lee, J.-H.; Kim, K.S.; Jeong, S.-M.; Kim, P.-H.; Chung, H.-T. Regulation of Neutrophil Adhesion by Pituitary Growth Hormone Accompanies Tyrosine Phosphorylation of Jak2, P125 FAK, and Paxillin. J. Immunol. 2000, 165, 2116–2123. [Google Scholar] [CrossRef] [Green Version]
- Milstien, S.; Katusic, Z. Oxidation of Tetrahydrobiopterin by Peroxynitrite: Implications for Vascular Endothelial Function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Borlak, J. Mechanistic Role of Cytochrome P450 Monooxygenases in Oxidized Low-Density Lipoprotein-Induced Vascular Injury: Therapy through LOX-1 Receptor Antagonism? Circ. Res. 2004, 94, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, E.; Grönbladh, A.; Zelleroth, S.; Diwakarla, S.; Nyberg, F.; Hallberg, M. Growth Hormone Is Protective against Acute Methadone-Induced Toxicity by Modulating the NMDA Receptor Complex. Neuroscience 2016, 339, 538–547. [Google Scholar] [CrossRef]
- Nylander, E.; Zelleroth, S.; Nyberg, F.; Grönbladh, A.; Hallberg, M. The Protective and Restorative Effects of Growth Hormone and Insulin-Like Growth Factor-1 on Methadone-Induced Toxicity In Vitro. Int. J. Mol. Sci. 2018, 19, 3627. [Google Scholar] [CrossRef] [Green Version]
- Erusalimsky, J.D. Vascular Endothelial Senescence: From Mechanisms to Pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Orosz, Z.; Labinskyy, N.; Rivera, A.; Xiangmin, Z.; Smith, K.; Csiszar, A. Increased Mitochondrial H2O2 Production Promotes Endothelial NF-KappaB Activation in Aged Rat Arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H37–H47. [Google Scholar] [CrossRef] [Green Version]
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear Factor-ΚB Activation Contributes to Vascular Endothelial Dysfunction via Oxidative Stress in Overweight/Obese Middle-Aged and Older Humans. Circulation 2009, 119, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Gautam, T.; Koncz, P.; Henthorn, J.C.; Pinto, J.T.; Ballabh, P.; Yan, H.; Mitschelen, M.; Farley, J.; Sonntag, W.E.; et al. Vasoprotective Effects of Life Span-Extending Peripubertal GH Replacement in Lewis Dwarf Rats. J. Gerontol.-Ser. A Biol. Sci. Med. Sci. 2010, 65A, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Sosnowska, D.; Podlutsky, A.; Koncz, P.; Sonntag, W.E.; Csiszar, A. Free Radical Production, Antioxidant Capacity, and Oxidative Stress Response Signatures in Fibroblasts from Lewis Dwarf Rats: Effects of Life Span-Extending Peripubertal GH Treatment. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66A, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Perret-Vivancos, C.; Abbate, A.; Ardail, D.; Raccurt, M.; Usson, Y.; Lobie, P.; Morel, G. Growth Hormone Activity in Mitochondria Depends on GH Receptor Box 1 and Involves Caveolar Pathway Targeting. Exp. Cell Res. 2005, 312, 215–232. [Google Scholar] [CrossRef]
- Ardail, D.; Debon, A.; Perret-Vivancos, C.; Biol-N’Garagba, M.-C.; Krantic, S.; Lobie, P.E.; Morel, G. Growth Hormone Internalization in Mitochondria Decreases Respiratory Chain Activity. Neuroendocrinology 2010, 91, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Perez, V.; Recchia, F.A.; Podlutsky, A.; Mukhopadhyay, P.; Losonczy, G.; Pacher, P.; Austad, S.N.; Bartke, A.; et al. Endothelial Function and Vascular Oxidative Stress in Long-Lived GH/IGF-Deficient Ames Dwarf Mice. AJP Hear. Circ. Physiol. 2008, 295, H1882–H1894. [Google Scholar] [CrossRef] [Green Version]
- Keane, J.; Tajouri, L.; Gray, B. Recombinant Human Growth Hormone and Insulin-Like Growth Factor-1 Do Not Affect Mitochondrial Derived Highly Reactive Oxygen Species Production in Peripheral Blood Mononuclear Cells under Conditions of Substrate Saturation In-Vitro. Nutr. Metab. (Lond.) 2016, 13, 45. [Google Scholar] [CrossRef] [Green Version]
- Friedenstein, A.J.; Petrakova, K.V.; Kurolesova, A.I.; Frolova, G.P. Heterotopic of Bone Marrow. Analysis of Precursor Cells for Osteogenic and Hematopoietic Tissues. Transplantation 1968, 6, 230–247. [Google Scholar] [CrossRef]
- da Silva Meirelles, L.; Fontes, A.M.; Covas, D.T.; Caplan, A.I. Mechanisms Involved in the Therapeutic Properties of Mesenchymal Stem Cells. Cytokine Growth Factor Rev. 2009, 20, 419–427. [Google Scholar] [CrossRef]
- Kassis, I.; Zangi, L.; Rivkin, R.; Levdansky, L.; Samuel, S.; Marx, G.; Gorodetsky, R. Isolation of Mesenchymal Stem Cells from G-CSF-Mobilized Human Peripheral Blood Using Fibrin Microbeads. Bone Marrow Transplant. 2006, 37, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Zhang, Y.; Hao, L.; Wang, F.; Liu, D.; Su, Y.; Sun, H. More Insight into Mesenchymal Stem Cells and Their Effects inside the Body. Expert Opin. Biol. Ther. 2010, 10, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Garcia, M.; Ning, H.; Banie, L.; Guo, Y.-L.; Lue, T.F.; Lin, C.-S. Defining Stem and Progenitor Cells within Adipose Tissue. Stem Cells Dev. 2008, 17, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Corselli, M.; Chen, C.-W.; Crisan, M.; Lazzari, L.; Péault, B. Perivascular Ancestors of Adult Multipotent Stem Cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1104–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siow, R. Migration of Adventitial Myofibroblasts Following Vascular Balloon Injury: Insights from In Vivo Gene Transfer to Rat Carotid Arteries. Cardiovasc. Res. 2003, 59, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Chen, S.-J.; Oparil, S.; Chen, Y.-F.; Thompson, J.A. Direct In Vivo Evidence Demonstrating Neointimal Migration of Adventitial Fibroblasts After Balloon Injury of Rat Carotid Arteries. Circulation 2000, 101, 1362–1365. [Google Scholar] [CrossRef] [Green Version]
- Faggin, E.; Puato, M.; Zardo, L.; Franch, R.; Millino, C.; Sarinella, F.; Pauletto, P.; Sartore, S.; Chiavegato, A. Smooth Muscle-Specific SM22 Protein Is Expressed in the Adventitial Cells of Balloon-Injured Rabbit Carotid Artery. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Haurani, M.; Pagano, P. Adventitial Fibroblast Reactive Oxygen Species as Autacrine and Paracrine Mediators of Remodeling: Bellwether for Vascular Disease? Cardiovasc. Res. 2007, 75, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.; Samee, S.; Chade, A.; Porcel, M.R.; Lerman, L.O.; Lerman, A. Differential Effect of Experimental Hypertension and Hypercholesterolemia on Adventitial Remodeling. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Davie, N.; Frid, M.; Gerasimovskaya, E.; Das, M. Role of the Adventitia in Pulmonary Vascular Remodeling. Physiology 2006, 21, 134–145. [Google Scholar] [CrossRef]
- Jin, K.; Li, B.; Lou, L.; Xu, Y.; Ye, X.; Yao, K.; Ye, J.; Gao, C. In Vivo Vascularization of MSC-Loaded Porous Hydroxyapatite Constructs Coated with VEGF-Functionalized Collagen/Heparin Multilayers. Sci. Rep. 2016, 6, 19871. [Google Scholar] [CrossRef] [Green Version]
- Khaki, M.; Salmanian, A.H.; Abtahi, H.; Ganji, A.; Mosayebi, G. Mesenchymal Stem Cells Differentiate to Endothelial Cells Using Recombinant Vascular Endothelial Growth Factor—A. Rep. Biochem. Mol. Biol. 2018, 6, 144–150. [Google Scholar] [PubMed]
- Devesa, J.; Caicedo, D. The Role of Growth Hormone on Ovarian Functioning and Ovarian Angiogenesis. Front. Endocrinol. (Lausanne) 2019, 10, 450. [Google Scholar] [CrossRef] [PubMed]
- Galvão, A.M.; Skarzynski, D.; Ferreira-Dias, G. Luteolysis and the Auto-, Paracrine Role of Cytokines from Tumor Necrosis Factor α and Transforming Growth Factor β Superfamilies. In Vitamins and Hormones; Academic Press: Cambridge, MA, USA, 2018; pp. 287–315. [Google Scholar] [CrossRef]
- Xie, Q.; Cheng, Z.; Chen, X.; Lobe, C.G.; Liu, J. The Role of Notch Signalling in Ovarian Angiogenesis. J. Ovarian Res. 2017, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, M.; Krawczynska, A.; Antushevinch, H.; Przemyslaw, A. Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction. Int. J. Mol. Sci. 2018, 19, 1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, W. Multiple Pathways Converge in the Development of a Collateral Circulation (Arteriogenesis). In Arteriogenesis—Molecular Regulation, Pathophysiology and Therapeutics I; Deindl, E., Schaper, W., Eds.; Shaker Verlag: Aachan, Germany, 2011; pp. 67–71. [Google Scholar]
- Brooks, A.J.; Waters, M.J. The Growth Hormone Receptor: Mechanism of Activation and Clinical Implications. Nat. Rev. Endocrinol. 2010, 6, 515–525. [Google Scholar] [CrossRef]
- Delafontaine, P.; Song, Y.H.; Li, Y. Expression, Regulation, and Function of IGF-1, IGF-1R, and IGF-1 Binding Proteins in Blood Vessels. Arterioscler. Thromb. Vasc. Biol. 2004, 435–444. [Google Scholar] [CrossRef]
- Lang, C.H.; Hong-Brown, L.; Frost, R.A. Cytokine Inhibition of JAK-STAT Signaling: A New Mechanism of Growth Hormone Resistance. Pediatr. Nephrol. 2005, 306–312. [Google Scholar] [CrossRef]
- Wickman, A.; Friberg, P.; Adams, M.A.; Matejka, G.L.; Brantsing, C.; Guron, G.; Isgaard, J. Induction of Growth Hormone Receptor and Insulin-Like Growth Factor-I MRNA in Aorta and Caval Vein during Hemodynamic Challenge. Hypertension 1997, 29, 123–130. [Google Scholar] [CrossRef]
- Owens, G.K.; Reidy, M.A. Hyperplastic Growth Response of Vascular Smooth Muscle Cells Following Induction of Acute Hypertension in Rats by Aortic Coarctation. Circ. Res. 1985, 57, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Delafontaine, P.; Lou, H.; Alexander, R.W. Regulation of Insulin-Like Growth Factor I Messenger RNA Levels in Vascular Smooth Muscle Cells. Hypertension 1991, 18, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Hansson, H.-A.; Jennische, E.; Skottner, A. IGF-I Expression in Blood Vessels Varies with Vascular Load. Acta Physiol. Scand. 1987, 129, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, S.W.; Yoshizato, H.; Barclay, J.L.; Brooks, A.J.; Behncken, S.N.; Kerr, L.M.; Millard, K.; Palethorpe, K.; Nielsen, K.; Clyde-Smith, J.; et al. An Agonist-Induced Conformational Change in the Growth Hormone Receptor Determines the Choice of Signalling Pathway. Nat. Cell Biol. 2008, 10, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Clapp, C.; Thebault, S.; Jeziorski, M.C.; Martínez De La Escalera, G. Peptide Hormone Regulation of Angiogenesis. Physiol. Rev. 2009, 89, 1177–1215. [Google Scholar] [CrossRef]
- Brüel, A.; Oxlund, H. Growth Hormone Influences the Content and Composition of Collagen in the Aorta from Old Rats. Mech. Ageing Dev. 2002, 123, 627–635. [Google Scholar] [CrossRef]
- Cittadini, A.; Strömer, H.; Katz, S.E.; Clark, R.; Moses, A.C.; Morgan, J.P.; Douglas, P.S. Differential Cardiac Effects of Growth Hormone and Insulin-Like Growth Factor-1 in the Rat. A Combined In Vivo and In Vitro Evaluation. Circulation 1996, 93, 800–809. [Google Scholar] [CrossRef]
- Le Roith, D.; Scavo, L.; Butler, A. What Is the Role of Circulating IGF-I? Trends Endocrinol. Metab. 2001, 12, 48–52. [Google Scholar] [CrossRef]
- Xu, X.Q.; Emerald, B.S.; Goh, E.L.K.; Kannan, N.; Miller, L.D.; Gluckman, P.D.; Liu, E.T.; Lobie, P.E. Gene Expression Profiling to Identify Oncogenic Determinants of Autocrine Human Growth Hormone in Human Mammary Carcinoma. J. Biol. Chem. 2005, 280, 23987–24003. [Google Scholar] [CrossRef] [Green Version]
- Nakonechnaya, A.O.; Jefferson, H.S.; Chen, X.; Shewchuk, B.M. Differential Effects of Exogenous and Autocrine Growth Hormone on LNCaP Prostate Cancer Cell Proliferation and Survival. J. Cell. Biochem. 2013, 114, 1322–1335. [Google Scholar] [CrossRef]
- Erman, A.; Wabitsch, M.; Goodyer, C.G. Human Growth Hormone Receptor (GHR) Expression in Obesity: II. Regulation of the Human GHR Gene by Obesity-Related Factors. Int. J. Obes. (Lond.) 2011, 35, 1520–1529. [Google Scholar] [CrossRef] [Green Version]
- Schwartzbauer, G.; Menon, R.K. Regulation of Growth Hormone Receptor Gene Expression. Mol. Genet. Metab. 1998, 63, 243–253. [Google Scholar] [CrossRef]
- Lobie, P.E.; Breipohl, W.; Aragón, J.G.; Waters, M.J. Cellular Localization of the Growth Hormone Receptor/Binding Protein in the Male and Female Reproductive Systems. Endocrinology 1990, 126, 2214–2221. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Corbacho, A.M.; Eiserich, J.P.; Garcia, C.; Lopez-Barrera, F.; Morales-Tlalpan, V.; Barajas-Espinosa, A.; Diaz-Muñoz, M.; Rubio, R.; Lin, S.-H.; et al. 16K-Prolactin Inhibits Activation of Endothelial Nitric Oxide Synthase, Intracellular Calcium Mobilization, and Endothelium-Dependent Vasorelaxation. Endocrinology 2004, 145, 5714–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govers, R.; Rabelink, T.J. Cellular Regulation of Endothelial Nitric Oxide Synthase. Am. J. Physiol. Renal Physiol. 2001, 280, F193–F206. [Google Scholar] [CrossRef] [PubMed]
- Walford, G.; Loscalzo, J. Nitric Oxide in Vascular Biology. J. Thromb. Haemost. 2003, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H. Nitric Oxide and the Mediation of the Hemodynamic Effects of Growth Hormone in Humans. J. Endocrinol. Investig. 1999, 22 (Suppl. 5), 75–81. [Google Scholar]
- Gonzalez, C.; Rosas-Hernandez, H.; Jurado-Manzano, B.; Ramirez-Lee, M.A.; Salazar-Garcia, S.; Martinez-Cuevas, P.P.; Velarde-Salcedo, A.J.; Morales-Loredo, H.; Espinosa-Tanguma, R.; Ali, S.F.; et al. The Prolactin Family Hormones Regulate Vascular Tone through NO and Prostacyclin Production in Isolated Rat Aortic Rings. Acta Pharmacol. Sin. 2015, 36, 572–586. [Google Scholar] [CrossRef] [Green Version]
- Campión, J.; Maestro, B.; Calle, C.; Dávila, N.; Barceló, B. Receptor de La Hormona de Crecimiento Humano: Características Estructurales, Estudio de Su Expresión y Regulación Génica. Endocrinol. Nutr. 1999, 46, 235–240. [Google Scholar]
- Caicedo, D.; Devesa, P.; Arce, V.M.; Requena, J.; Devesa, J. Chronic Limb-Threatening Ischemia Could Benefit from Growth Hormone Therapy for Wound Healing and Limb Salvage. Ther. Adv. Cardiovasc. Dis. 2017, 1–20. [Google Scholar] [CrossRef]
- Heil, M.; Schaper, W. Insights into Pathways of Arteriogenesis. Curr. Pharm. Biotechnol. 2007, 8, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Meazza, C.; Pagani, S.; Travaglino, P.; Bozzola, M. Effect of Growth Hormone (GH) on the Immune System. Pediatr. Endocrinol. Rev. 2004, 1, 490–495. [Google Scholar] [PubMed]
- Fasshauer, M.; Klein, J.; Kralisch, S.; Klier, M.; Lossner, U.; Bluher, M.; Paschke, R. Monocyte Chemoattractant Protein 1 Expression Is Stimulated by Growth Hormone and Interleukin-6 in 3T3-L1 Adipocytes. Biochem. Biophys. Res. Commun. 2004, 317, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, A.; Piek, J.; Van Royen, N. Collateral Artery Growth in Man, from Assesment to Stimulation. In Arteriogenesis—Molecular Regulation, Pathophysiology and Therapeutics I; Deindl, E., Schaper, W., Eds.; Shaker Verlag: Aachen, Germany, 2011; pp. 167–175. [Google Scholar]
- Shireman, P.K.; Contreras-Shannon, V.; Reyes-Reyna, S.M.; Robinson, S.C.; McManus, L.M. MCP-1 Parallels Inflammatory and Regenerative Responses in Ischemic Muscle. J. Surg. Res. 2006, 134, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.K.; Fisker, S.; Dall, R.; Ledet, T.; Jørgensen, J.O.L.; Rasmussen, L.M. Growth Hormone Increases Vascular Cell Adhesion Molecule 1 Expression: In Vivo and In Vitro Evidence. J. Clin. Endocrinol. Metab. 2004, 89, 909–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Yu, W.-Y.; Yu, D.-J.; Zhao, T.-L.; Wu, L.-J.; Han, W.-Y. The Effects of Recombinant Human Growth Hormone (RHGH) on Survival of Slender Narrow Pedicle Flap and Expressions of Vascular Endothelial Growth Factor (VEGF) and Classification Determinant 34 (CD34). Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 771–777. [Google Scholar] [CrossRef]
- Nielsen, J.S.; McNagny, K.M. Novel Functions of the CD34 Family. J. Cell Sci. 2008, 121, 3683–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Klaauw, A.A.; Pereira, A.M.; Rabelink, T.J.; Corssmit, E.P.M.; Zonneveld, A.-J.; Pijl, H.; de Boer, H.C.; Smit, J.W.A.; Romijn, J.A.; de Koning, E.J.P. Recombinant Human GH Replacement Increases CD34+ Cells and Improves Endothelial Function in Adults with GH Deficiency. Eur. J. Endocrinol. 2008, 159, 105–111. [Google Scholar] [CrossRef]
- Kiess, W.; Butenandt, O. Specific Growth Hormone Receptors on Human Peripheral Mononuclear Cells: Reexpression, Identification, and Characterization. J. Clin. Endocrinol. Metab. 1985, 60, 740–746. [Google Scholar] [CrossRef]
- Bresson, J.L.; Jeay, S.; Gagnerault, M.C.; Kayser, C.; Beressi, N.; Wu, Z.; Kinet, S.; Dardenne, M.; Postel-Vinay, M.C. Growth Hormone (GH) and Prolactin Receptors in Human Peripheral Blood Mononuclear Cells: Relation with Age and GH-Binding Protein. Endocrinology 1999, 140, 3203–3209. [Google Scholar] [CrossRef]
- Hattori, N. Expression, Regulation and Biological Actions of Growth Hormone (GH) and Ghrelin in the Immune System. Growth Horm. IGF Res. 2009, 19, 187–197. [Google Scholar] [CrossRef]
- Stuart, C.; Meehan, R.; Neale, L.; Cintron, N.; Furlanetto, R. Insulin-Like Growth Factor-I Binds Selectively to Human Peripheral Blood Monocytes and B-Lymphocytes*. J. Clin. Endocrinol. Metab. 1991, 72, 1117–1122. [Google Scholar] [CrossRef]
- Arkins, S.; Rebeiz, N.; Biragyn, A.; Reese, D.L.; Kelley, K.W. Murine Macrophages Express Abundant Insulin-Like Growth Factor-I Class I Ea and Eb Transcripts. Endocrinology 1993, 133, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, S.; Castier, Y.; Tedgui, A. Molecular Mechanisms of the Vascular Responses to Haemodynamic Forces. J. Intern. Med. 2006, 259, 381–392. [Google Scholar] [CrossRef]
- Yang, H. Effect of Aging on Angiogenesis and Arteriogenesis. Curr. Cardiol. Rev. 2007, 3, 65–74. [Google Scholar] [CrossRef]
- Hellingman, A.; Seghers, L.; Quax, P.; Van Weel, V. Bone Marrow Derived Cells in Artriogenesis: A Crucial Role for Leukocytes. In Arteriogenesis—Molecular Regulation, Pathophysiology and Therapeutics I; Deindl, E., Scahper, W., Eds.; Shaker Verlag: Aachen, Germany, 2011; pp. 145–150. [Google Scholar]
- Wiedermann, C.J.; Reinisch, N.; Kähler, C.; Geisen, F.; Zilian, U.; Herold, M.; Braunsteiner, H. In Vivo Activation of Circulating Monocytes by Exogenous Growth Hormone in Man. Brain Behav. Immun. 1992, 6, 387–393. [Google Scholar] [CrossRef]
- Zhu, T.; Goh, E.L.K.; LeRoith, D.; Lobie, P.E. Growth Hormone Stimulates the Formation of a Multiprotein Signaling Complex Involving P130(Cas) and CrkII: Resultant Activation of c-Jun N-Terminal Kinase/Stress-Activated Protein Kinase (JNK/SAPK). J. Biol. Chem. 1998, 273, 33864–33875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Ouchi, T.; Hanafusa, H. Downstream of Crk Adaptor Signaling Pathway: Activation of Jun Kinase by v-Crk through the Guanine Nucleotide Exchange Protein C3G. Proc. Natl. Acad. Sci. USA 1997, 94, 2356–2361. [Google Scholar] [CrossRef] [Green Version]
- Wiktor-Jedrzejczak, W.; Urbanowska, E.; Aukerman, S.L.; Pollard, J.W.; Stanley, E.R.; Ralph, P.; Ansari, A.A.; Sell, K.W.; Szperl, M. Correction by CSF-1 of Defects in the Osteopetrotic Op/Op Mouse Suggests Local, Developmental, and Humoral Requirements for This Growth Factor. Exp. Hematol. 1991, 19, 1049–1054. [Google Scholar]
- Baxter, J.; Blalock, J.; Weigent, D. Characterization of Immunoreactive Insulin-Like Growth Factor-I from Leukocytes and Its Regulation by Growth Hormone*. Endocrinology 1991, 129, 1727–1734. [Google Scholar] [CrossRef]
- Gow, D.J.; Sester, D.P.; Hume, D.A. CSF-1, IGF-1, and the Control of Postnatal Growth and Development. J. Leukoc. Biol. 2010, 88, 475–481. [Google Scholar] [CrossRef]
- Hume, D.A.; Halpin, D.; Charlton, H.; Gordon, S. The Mononuclear Phagocyte System of the Mouse Defined by Immunohistochemical Localization of Antigen F4/80: Macrophages of Endocrine Organs. Proc. Natl. Acad. Sci. USA 1984, 81, 4174–4177. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Hoffman, A.; Yang, Y.; Yan, J.; Tie, G.; Bagshahi, H.; Nowicki, P.T.; Messina, L.M. Endothelial Nitric Oxide Synthase Affects Both Early and Late Collateral Arterial Adaptation and Blood Flow Recovery after Induction of Hind Limb Ischemia in Mice. J. Vasc. Surg. 2010, 51, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, D.; Tamay, F.; Álvarez, S.; Mendieta, J. Current Advances in the Biochemical and Physiological Aspects of the Treatment of Type 2 Diabetes Mellitus with Thiazolidinediones. PPAR Res. 2016, 2016, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Prior, B.M.; Lloyd, P.G.; Ren, J.; Li, H.; Yang, H.T.; Laughlin, M.H.; Terjung, R.L. Time Course of Changes in Collateral Blood Flow and Isolated Vessel Size and Gene Expression after Femoral Artery Occlusion in Rats. Am. J. Physiol. Circ. Physiol. 2004, 287, H2434–H2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mees, B.; Wagner, S.; Ninci, E.; Tribulova, S.; Martin, S.; Van Haperen, R.; Kostin, S.; Heil, M.; De Crom, R.; Schaper, W. Endothelial Nitric Oxide Synthase Activity Is Essential for Vasodilation during Blood Flow Recovery but Not for Arteriogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1926–1933. [Google Scholar] [CrossRef] [Green Version]
- Unthank, J.; Haas, T.L.; Miller, S. Impact of Shear Level and Cardiovascular Risk Factors on Bioavailable Nitric Oxide and Outward Vascular Remodeling in Mesenteric Arteries. In Arteriogenesis-Molecular Regulation, Pathophysiology, and Therapeutics I; Shaker Verlag Publishing: Aachen, Germany, 2011; No. May 2014; pp. 89–119. [Google Scholar]
- Stasch, J.P.; Schmidt, P.M.; Nedvetsky, P.I.; Nedvetskaya, T.Y.; Arun Kumar, H.S.; Meurer, S.; Deile, M.; Taye, A.; Knorr, A.; Lapp, H.; et al. Targeting the Heme-Oxidized Nitric Oxide Receptor for Selective Vasodilatation of Diseased Blood Vessels. J. Clin. Investig. 2006, 116, 2552–2561. [Google Scholar] [CrossRef] [Green Version]
- Gladwin, M.T. Deconstructing Endothelial Dysfunction: Soluble Guanylyl Cyclase Oxidation and the NO Resistance Syndrome. J. Clin. Investig. 2006, 116, 2330–2332. [Google Scholar] [CrossRef]
- Bauersachs, J.; Bouloumié, A.; Mülsch, A.; Wiemer, G.; Fleming, I.; Busse, R. Vasodilator Dysfunction in Aged Spontaneously Hypertensive Rats: Changes in NO Synthase III and Soluble Guanylyl Cyclase Expression, and in Superoxide Anion Production. Cardiovasc. Res. 1998, 37, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Bir, S.C.; Kolluru, G.K.; Fang, K.; Kevil, C.G. Redox Balance Dynamically Regulates Vascular Growth and Remodeling. In Seminars in Cell and Developmental Biology; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 745–757. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.W.; Parker, D.E.; Montgomery, P.S.; Sosnowska, D.; Casanegra, A.I.; Esponda, O.L.; Ungvari, Z.; Csiszar, A.; Sonntag, W.E. Impaired Vascular Endothelial Growth Factor A and Inflammation in Patients with Peripheral Artery Disease. Angiology 2014, 65, 683–690. [Google Scholar] [CrossRef]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho Suppresses TNF-Alpha-Induced Expression of Adhesion Molecules in the Endothelium and Attenuates NF-KappaB Activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef]
- Yi, C.; Cao, Y.; Mao, S.H.; Liu, H.; Ji, L.L.; Xu, S.Y.; Zhang, M.; Huang, Y. Recombinant Human Growth Hormone Improves Survival and Protects against Acute Lung Injury in Murine Staphylococcus Aureus Sepsis. Inflamm. Res. 2009, 58, 855–862. [Google Scholar] [CrossRef]
- Adamopoulos, S. Effects of Growth Hormone on Circulating Cytokine Network, and Left Ventricular Contractile Performance and Geometry in Patients with Idiopathic Dilated Cardiomyopathy. Eur. Heart J. 2003, 24, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Masternak, M.M.; Bartke, A. Growth Hormone, Inflammation and Aging. Pathobiol. Aging Age-Relat. Dis. 2012, 2, 17293. [Google Scholar] [CrossRef]
- Rentrop, K.P.; Feit, F.; Sherman, W.; Thornton, J.C. Serial Angiographic Assessment of Coronary Artery Obstruction and Collateral Flow in Acute Myocardial Infarction. Report from the Second Mount Sinai-New York University Reperfusion Trial. Circulation 1989, 80, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Piek, J.J.; van Liebergen, R.A.; Koch, K.T.; Peters, R.J.G.; David, G.K. Clinical, Angiographic and Hemodynamic Predictors of Recruitable Collateral Flow Assessed During Balloon Angioplasty Coronary Occlusion. J. Am. Coll. Cardiol. 1997, 29, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Tang, G.; Yan, J.; Park, B.; Hoffman, A.; Tie, G.; Messina, L.M. Cellular and Molecular Mechanism Regulating Blood Flow Recovery in Acute versus Gradual Femoral Artery Occlusion Are Distinct in the Mouse. J. Vasc. Surg. 2009, 48, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovsky, E.D.; Draghia-Akli, R. Insulin-Like Growth Factor I Plasmid Therapy Promotes In Vivo Angiogenesis. Mol. Ther. 2004, 9, 46–55. [Google Scholar] [CrossRef]
- Kusano, K.; Tsutsumi, Y.; Dean, J.; Gavin, M.; Ma, H.; Silver, M.; Thorne, T.; Zhu, Y.; Losordo, D.W.; Aikawa, R. Long-Term Stable Expression of Human Growth Hormone by RAAV Promotes Myocardial Protection Post-Myocardial Infarction. J. Mol. Cell. Cardiol. 2007, 42, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Dobruckia, L.W.; Tsutsumib, Y.; Kalinowskia, L.; Deanb, J.; Gavinb, M.; Senb, S.; Mendizabald, M.; Sinusasa, A.J.; Aikawab, R. Analysis of Angiogenesis Induced by Local IGF-1 Expression after Myocardial Infarction Using MicroSPECT-CT Imaging. J. Mol. Cell. Cardiol. 2009, 6, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Pelisek, J.; Shimizu, M.; Nikol, S. Differential Developmental Origin of Arteries: Impact on Angiogenesis and Arteriogenesis. Med. Chem. Rev.-Online 2004, 1, 317–326. [Google Scholar] [CrossRef]
- Palmer-Kazen, U.; Wariaro, D.; Luo, F.; Wahlberg, E. Vascular Endothelial Cell Growth Factor and Fibroblast Growth Factor 2 Expression in Patients with Critical Limb Ischemia. J. Vasc. Surg. 2004, 39, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Rissanen, T.T.; Vajanto, I.; Hiltunen, M.O.; Rutanen, J.; Kettunen, M.I.; Niemi, M.; Leppänen, P.; Turunen, M.P.; Markkanen, J.E.; Arve, K.; et al. Expression of Vascular Endothelial Growth Factor and Vascular Endothelial Growth Factor Receptor-2 (KDR/Flk-1) in Ischemic Skeletal Muscle and Its Regeneration. Am. J. Pathol. 2002, 160, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Klinnikova, M.G.; Bakarev, M.A.; Nikityuk, D.B.; Lushnikova, E.L. Immunohistochemical Study of the Expression of Vascular Endothelial Growth Factor Receptor-2 (KDR/Flk-1) during Myocardial Infarction. Bull. Exp. Biol. Med. 2017, 163, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Lasch, M.; Kleinert, E.C.; Meister, S.; Kumaraswami, K.; Buchheim, J.-I.; Grantzow, T.; Lautz, T.; Salpisti, S.; Fischer, S.; Troidl, K.; et al. Extracellular RNA Released Due to Shear Stress Controls Natural Bypass Growth by Mediating Mechanotransduction in Mice. Blood 2019, 134, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.L.; Sessa, W.C. ENOS-Derived Nitric Oxide Regulates Endothelial Barrier Function through VE-Cadherin and Rho GTPases. J. Cell Sci. 2013, 126 (Pt 24), 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, J.M.; Lee, E.J. Functional Expression of CXCR4 in Somatotrophs: CXCL12 Activates GH Gene, GH Production and Secretion, and Cellular Proliferation. J. Endocrinol. 2008, 199, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, F.; Bajetto, A.; Porcile, C.; Pattarozzi, A.; Schettini, G.; Florio, T. Role of Stromal Cell-Derived Factor 1 (SDF1/CXCL12) in Regulating Anterior Pituitary Function. J. Mol. Endocrinol. 2007, 38, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Smaniotto, S.; Martins-Neto, A.A.; Dardenne, M.; Savino, W. Growth Hormone Is a Modulator of Lymphocyte Migration. Neuroimmunomodulation 2011, 18, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Bolamperti, S.; Guidobono, F.; Rubinacci, A.; Villa, I. The Role of Growth Hormone in Mesenchymal Stem Cell Commitment. Int. J. Mol. Sci. 2019, 20, 5264. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Adesanya, T.M.A.; Zhang, L.; Xie, N.; Chen, Z.; Fu, M.; Zhang, J.; Zhang, J.; Tan, T.; Kilic, A.; et al. Delivery of Placenta-Derived Mesenchymal Stem Cells Ameliorates Ischemia Induced Limb Injury by Immunomodulation. Cell. Physiol. Biochem. 2014, 34, 1998–2006. [Google Scholar] [CrossRef] [Green Version]
- Katare, R.; Riu, F.; Rowlinson, J.; Lewis, A.; Holden, R.; Meloni, M.; Reni, C.; Wallrapp, C.; Emanueli, C.; Madeddu, P. Perivascular Delivery of Encapsulated Mesenchymal Stem Cells Improves Postischemic Angiogenesis Via Paracrine Activation of VEGF-A. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1872–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurila, J.P.; Laatikainen, L.; Castellone, M.D.; Trivedi, P.; Heikkila, J.; Hinkkanen, A.; Hematti, P.; Laukkanen, M.O. Human Embryonic Stem Cell-Derived Mesenchymal Stromal Cell Transplantation in a Rat Hind Limb Injury Model. Cytotherapy 2009, 11, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Galderisi, U.; Marino, I.R. From the Laboratory Bench to the Patient’s Bedside: An Update on Clinical Trials with Mesenchymal Stem Cells. J. Cell. Physiol. 2007, 211, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Werther, G.A.; Haynes, K.; Waters, M.J. Growth Hormone (GH) Receptors Are Expressed on Human Fetal Mesenchymal Tissues—Identification of Messenger Ribonucleic Acid and GH-Binding Protein. J. Clin. Endocrinol. Metab. 1993, 76, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Noiseux, N.; Gnecchi, M.; Lopez-Ilasaca, M.; Zhang, L.; Solomon, S.D.; Deb, A.; Dzau, V.J.; Pratt, R.E. Mesenchymal Stem Cells Overexpressing Akt Dramatically Repair Infarcted Myocardium and Improve Cardiac Function despite Infrequent Cellular Fusion or Differentiation. Mol. Ther. 2006, 14, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Olarescu, N.C.; Berryman, D.E.; Householder, L.A.; Lubbers, E.R.; List, E.O.; Benencia, F.; Kopchick, J.J.; Bollerslev, J. GH Action Influences Adipogenesis of Mouse Adipose Tissue-Derived Mesenchymal Stem Cells. J. Endocrinol. 2015, 226, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Bolamperti, S.; Signo, M.; Spinello, A.; Moro, G.; Fraschini, G.; Guidobono, F.; Rubinacci, A.; Villa, I. GH Prevents Adipogenic Differentiation of Mesenchymal Stromal Stem Cells Derived from Human Trabecular Bone via Canonical Wnt Signaling. Bone 2018, 112, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ito, W.; Fleming, I.; Deindl, E.; Sauer, A.; Wiesnet, M.; Busse, R.; Schaper, J.; Schaper, W. Ultrastructure and Molecular Histology of Rabbit Hind-Limb Collateral Artery Growth (Arteriogenesis). Virchows Arch. 2000, 436, 257–270. [Google Scholar] [CrossRef]
- Messias de Lima, C.F.; Dos Santos Reis, M.D.; da Silva Ramos, F.W.; Ayres-Martins, S.; Smaniotto, S. Growth Hormone Modulates In Vitro Endothelial Cell Migration and Formation of Capillary-Like Structures. Cell Biol. Int. 2017, 41, 577–584. [Google Scholar] [CrossRef]
- Erikstrup, C.; Pedersen, L.M.; Heickendorff, L.; Ledet, T.; Rasmussen, L.M. Production of Hyaluronan and Chondroitin Sulphate Proteoglycans from Human Arterial Smooth Muscle—The Effect of Glucose, Insulin, IGF-I or Growth Hormone. Eur. J. Endocrinol. 2001, 145, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Cen, Y.; Liu, J.; Qin, Y.; Liu, R.; Wang, H.; Zhou, Y.; Wang, S.; Hu, Z. Denervation in Femoral Artery-Ligated Hindlimbs Diminishes Ischemic Recovery Primarily via Impaired Arteriogenesis. PLoS ONE 2016, 11, e0154941. [Google Scholar] [CrossRef] [PubMed]
- Sverrisdottir, Y.B.; Elam, M.; Herlitz, H.; Bengtsson, B.A.; Johannsson, G. Intense Sympathetic Nerve Activity in Adults with Hypopituitarism and Untreated Growth Hormone Deficiency. J. Clin. Endocrinol. Metab. 1998, 83, 1881–1885. [Google Scholar] [CrossRef]
- Sverrisdóttir, Y.B.; Elam, M.; Caidahl, K.; Söderling, A.-S.; Herlitz, H.; Johannsson, G. The Effect of Growth Hormone (GH) Replacement Therapy on Sympathetic Nerve Hyperactivity in Hypopituitary Adults: A Double-Blind, Placebo-Controlled, Crossover, Short-Term Trial Followed by Long-Term Open GH Replacement in Hypopituitary Adults. J. Hypertens. 2003, 21, 1905–1914. [Google Scholar] [CrossRef]
- Martínez-Nieves, B.; Dunbar, J.C. Vascular Dilatatory Responses to Sodium Nitroprusside (SNP) and Alpha-Adrenergic Antagonism in Female and Male Normal and Diabetic Rats. Proc. Soc. Exp. Biol. Med. 1999, 222, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, M.S.; van Golde, J.M.; Schaper, N.C.; Stehouwer, C.D.; Huijberts, M.S. Diabetes Impairs Arteriogenesis in the Peripheral Circulation: Review of Molecular Mechanisms. Clin. Sci. (Lond.) 2010, 119, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meusel, M.; Herrmann, M.; Machleidt, F.; Franzen, K.F.; Krapalis, A.F.; Sayk, F. GHRH-Mediated GH Release Is Associated with Sympathoactivation and Baroreflex Resetting: A Microneurographic Study in Healthy Humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R15–R24. [Google Scholar] [CrossRef]
- Lautz, T.; Lasch, M.; Borgolte, J.; Troidl, K.; Pagel, J.-I.; Caballero-Martinez, A.; Kleinert, E.C.; Walzog, B.; Deindl, E. Midkine Controls Arteriogenesis by Regulating the Bioavailability of Vascular Endothelial Growth Factor A and the Expression of Nitric Oxide Synthase 1 and 3. EBioMedicine 2018, 27, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Maliza, R.; Tofrizal, A.; Batchuluun, K.; Ramadhani, D.; Tsukada, T.; Azuma, M.; Horiguchi, K.; Kikuchi, M.; Yashiro, T. In Situ Hybridization Analysis of the Temporospatial Expression of the Midkine/Pleiotrophin Family in Rat Embryonic Pituitary Gland. Cell Tissue Res. 2014, 357, 337–344. [Google Scholar] [CrossRef]
- Fujiwara, K.; Horiguchi, K.; Maliza, R.; Tofrizal, A.; Batchuluun, K.; Ramadhani, D.; Syaidah, R.; Tsukada, T.; Azuma, M.; Kikuchi, M.; et al. Expression of the Heparin-Binding Growth Factor Midkine and Its Receptor, Ptprz1, in Adult Rat Pituitary. Cell Tissue Res. 2015, 359, 909–914. [Google Scholar] [CrossRef]
- Mulvany, M. Vascular Remodelling of Resistance Vessels: Can We Define This? Cardiovasc. Res. 1999, 41, 9–13. [Google Scholar] [CrossRef]
- Weckbach, L.; Preissner, K.; Deindl, E. The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation. Int. J. Mol. Sci. 2018, 19, 2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.; Neidert, M.C.; Tschopp, O.; Sze, L.; Bernays, R.L. Growth Hormone and Klotho. J. Endocrinol. 2013, 219, R37–R57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the Mouse Klotho Gene Leads to a Syndrome Resembling Ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Z. Molecular Basis of Klotho: From Gene to Function in Aging. Endocr. Rev. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinek, T.; Modan-Moses, D. Klotho and the Growth Hormone/Insulin-Like Growth Factor 1 Axis: Novel Insights into Complex Interactions. In Vitamins and Hormones; Academic Press: Cambridge, MA, USA, 2016; Volume 101, pp. 85–118. [Google Scholar] [CrossRef]
- Chung, C.-P.; Chang, Y.-C.; Ding, Y.; Lim, K.; Liu, Q.; Zhu, L.; Zhang, W.; Lu, T.-S.; Molostvov, G.; Zehnder, D.; et al. α-Klotho Expression Determines Nitric Oxide Synthesis in Response to FGF-23 in Human Aortic Endothelial Cells. PLoS ONE 2017, 12, e0176817. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H. Suppression of Aging in Mice by the Hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Kusaba, T.; Okigaki, M.; Matui, A.; Murakami, M.; Ishikawa, K.; Kimura, T.; Sonomura, K.; Adachi, Y.; Shibuya, M.; Shirayama, T.; et al. Klotho Is Associated with VEGF Receptor-2 and the Transient Receptor Potential Canonical-1 Ca2+ Channel to Maintain Endothelial Integrity. Proc. Natl. Acad. Sci. USA 2010, 107, 19308–19313. [Google Scholar] [CrossRef] [Green Version]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, Acute Effects of Klotho and FGF23 on Vascular Smooth Muscle and Endothelium. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrolled Patients | Sex | Age (Mean ± SD) | Comorbidity | Baseline ABI (Mean) | Rutherford class 5–6 |
|---|---|---|---|---|---|
| 36 | Male: 28 | 71 ± 12.7 | Heart disease:47% | 0.19 | 70.6% |
| Female:8 | DM:59% | ||||
| Neuropathy:57.6% | |||||
| CRF:26.5% | |||||
| Therapy | Dose | Duration of treatment | Follow-up | Follow-up Periods | |
| rhGH vs. Placebo | 0.4 mg/day | 8 weeks | 12 months | 0–2; 3–6; 7–12 months |
| Total | GH Group | Placebo Group | p Value | |||||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | p | ||
| Age | < 65 65–80 > 80 | 14 8 12 | 41.18 23.53 35.29 | 6 3 9 | 33.33 16.67 50 | 8 5 3 | 50 31.25 18.75 | 0.1211 |
| Gender | Male Female | 27 7 | 79.41 20.59 | 13 5 | 72.22 27.78 | 14 2 | 87.5 12.5 | 0.2715 |
| Etiology | Atherosclerosis Buerger Scleroderma Mix | 24 3 1 6 | 70.59 8.82 2.94 17.65 | 11 1 1 5 | 61.11 5.56 5.56 27.78 | 13 2 - 1 | 81.25 12.5 - 6.25 | |
| HT | No Yes | 12 22 | 35.29 64.71 | 4 14 | 22.22 77.78 | 8 8 | 50 50 | 0.0907 |
| DM | No Yes | 14 20 | 41.18 58.82 | 6 12 | 33.33 66.67 | 8 8 | 50 50 | 0.3243 |
| CRF | No Yes | 25 9 | 73.53 26.47 | 13 5 | 72.22 27.78 | 12 4 | 75 25 | 0.8546 |
| HD | No Yes | 18 16 | 52.94 47.06 | 8 10 | 44.44 55.56 | 10 6 | 62.5 37.5 | 0.2924 |
| Dialysis | No Yes | 33 1 | 97.06 2.94 | 17 1 | 94.44 5.56 | 16 - | 100 - | 0.3386 |
| Tobacco | No Ex-smoker <1y Smoker | 24 2 8 | 70.59 5.88 23.53 | 16 1 1 | 88.89 5.56 5.56 | 8 1 7 | 50 6.25 43.75 | 0.0107 |
| Rutherford | 3 4 5 6 | 5 5 15 9 | 14.71 14.71 44.12 26.47 | 3 - 9 6 | 16.67 - 50 33.33 | 2 5 6 3 | 12.5 31.25 37.5 18.75 | 0.188 |
| Rest Pain | No Yes | 8 26 | 23.53 76.47 | 5 13 | 27.78 72.22 | 3 13 | 18.75 81.25 | 0.5356 |
| Trophic Lesion | No Yes | 11 23 | 32.35 67.65 | 3 15 | 16.67 83.33 | 8 3 | 50 50 | 0.0381 |
| Neuropathy | No Yes | 14 19 | 42.42 57.58 | 7 10 | 41.18 58.82 | 7 9 | 43.75 56.25 | 0.8812 |
| GH-Treated | Placebo | ||||||
|---|---|---|---|---|---|---|---|
| Obs. | Mean | SD | Obs. | Mean | SD | p Value | |
| TNF -α Baseline | 16 | 12.35 | 5.2 | 16 | 8.78 | 3.94 | 0.018 |
| TNF -α Final | 15 | 10.92 | 5.12 | 14 | 8.04 | 3.6 | 0.046 |
| CRP Baseline | 16 | 2.07 | 2.86 | 16 | 0.78 | 0.69 | 0.045 |
| CRP Final | 15 | 1.1 | 1.34 | 14 | 3.42 | 7.51 | 0.218 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caicedo, D.; Devesa, P.; Alvarez, C.V.; Devesa, J. Why Should Growth Hormone (GH) Be Considered a Promising Therapeutic Agent for Arteriogenesis? Insights from the GHAS Trial. Cells 2020, 9, 807. https://doi.org/10.3390/cells9040807
Caicedo D, Devesa P, Alvarez CV, Devesa J. Why Should Growth Hormone (GH) Be Considered a Promising Therapeutic Agent for Arteriogenesis? Insights from the GHAS Trial. Cells. 2020; 9(4):807. https://doi.org/10.3390/cells9040807
Chicago/Turabian StyleCaicedo, Diego, Pablo Devesa, Clara V. Alvarez, and Jesús Devesa. 2020. "Why Should Growth Hormone (GH) Be Considered a Promising Therapeutic Agent for Arteriogenesis? Insights from the GHAS Trial" Cells 9, no. 4: 807. https://doi.org/10.3390/cells9040807
APA StyleCaicedo, D., Devesa, P., Alvarez, C. V., & Devesa, J. (2020). Why Should Growth Hormone (GH) Be Considered a Promising Therapeutic Agent for Arteriogenesis? Insights from the GHAS Trial. Cells, 9(4), 807. https://doi.org/10.3390/cells9040807