1. Introduction
Pancreatic cancer is the seventh leading cause of cancer-related deaths globally; it accounts for more than 20% of all deaths caused by abdominal cancers and approximately 5% of all cancer-related deaths [
1]. Most deaths are histologically attributed to the pancreatic ductal adenocarcinoma (PDAC) subtype, which originates from the pancreatic duct epithelium, represents approximately 85% of the total incidence of pancreatic cancer, and is often unresectable; consequently, patients have limited treatment options and exhibit high morbidity and mortality rates [
1,
2]. A yearly increase was reported in PDAC-specific mortality rates (approximately 98% mortality/incidence), and a consistently low five-year survival rate of <5% was reported over the past decade [
1].
Currently, the standard of care is gemcitabine with nanoparticle albumin-bound paclitaxel, erlotinib, or capecitabine, while for advanced stage (III and IV) or metastatic PDAC, the preferred chemotherapy is a combination of fluorouracil, leucovorin, irinotecan, and oxaliplatin, known as FOLFIRINOX chemotherapy; however, these chemotherapeutic drugs are associated with a high risk of severe drug-related toxicity, acquired resistance, and non-significant survival benefits [
3,
4]. Hence, the discovery of new actionable molecular oncotargets is necessary. Furthermore, screening methods should be developed for identifying novel and highly efficacious therapeutic agents that inhibit disease progression, alleviate resistance to treatment, and improve prognosis in patients with PDAC.
The involvement of highly conserved and ubiquitous transmembrane tyrosine kinase receptors, namely fibroblast growth factor receptors (FGFRs), in tumorigenesis, cell fate determination, survival, motility, angiogenesis, and malignantization of tumor cells, as well as in reduced sensitivity to anticancer therapeutics and poor prognosis is extensively documented [
5]. Notably, genetic alterations in FGFR1, such as amplification or an increase in gene copy number, are positively correlated with overexpression and are more prevalent than genetic aberrations in FGFR2, FGFR3, and FGFR4 [
5,
6]. Hence, Lehnen et al., studying the role of the FGFR1 gene copy number and expression pattern in patients with PDAC, suggested that the association between FGFR1 amplification, mRNA or protein expression, and the proliferative potential of PDAC cells can be exploited for therapeutic purposes using FGFR1 inhibitors [
7]. This is consistent with results indicating that enhanced FGFR signaling predisposed pancreatic cancer cells to the killing effect of dovitinib, a potent FGFR inhibitor, in preclinical models [
8], thus indicating that the pharmacological or molecular targeting of FGFR1 in patients with PDAC has therapeutic potential. However, despite the actual or predicted therapeutic benefits of FGFR1-targeting therapeutics, the post-treatment development of resistance due to accrue de novo genetic alterations in the kinase targets of these therapeutics following extended exposure to FGFR1 inhibitors is extensively documented [
9,
10,
11]. Therefore, the discovery or development of adjuvant or maintenance therapeutic agents for treating patients with PDAC harboring FGFR1 aberrations is necessary.
Poly (ADP ribose) polymerase (PARP) plays a critical role in the sensing of genomic damage signals, DNA repair and replication, inflammation, transcriptional and posttranscriptional gene expression modulation, and cell death regulation. PARP has also been implicated in several pathological processes, including carcinogenesis, by PARylation, direct or mediated interactions with oncogenes, and regulation of oncogenic transcription factors [
12]; thus the increased interest in and exploration of the therapeutic potential and clinical feasibility of PARP inhibition as an effective anticancer strategy in the last decade. Several PARP inhibitors are being developed or are currently undergoing clinical evaluation. As the concept of synthetic lethality in anticancer therapy continues to gain traction, the probable exploitability of the DNA repair-disrupting potential of PARP inhibitors (PARPi) is receiving a second look. In the context of the present study, synthetic lethality is implied if the impairment of either of two oncogenic pathways is not lethal or sufficiently detrimental to the viability of cancerous cells, but their combination induces lethality in the cancerous cells. An increasing amount of preclinical and clinical evidence suggests that PARPi may potentiate the effect of conventional chemotherapeutics, enhance sensitivity to radiation therapy, and serve as adjuvants or maintenance therapeutic agents [
12].
Against the background of the increasing incidence of acquired resistance to the FGFR1 inhibitor and our previous work that partially demonstrated the caspase-3/PARP-mediated anticancer and anti-metastasis efficacy of PD173074, a selective FGFR1 inhibitor, against aldehyde dehydrogenase (ALDH)-rich or FGFR1-high PDAC cells [
13], the present study investigates the probable synthetic lethality and therapeutic efficacy of targeted combinatorial FGFR1/PARP inhibition in patients with PDAC harboring FGFR1 aberrations using a multifaceted approach, including bioinformatics-aided prediction and visualization, drug screening, molecular biology, and functional assays.
2. Materials and Methods
2.1. Analysis of Public Cancer Datasets
Public online cancer databases, namely The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and Broad Institute Cancer Cell Line Encyclopedia, were used in this study. We probed the provisional TCGA pancreatic adenocarcinoma (TCGA PAAD) Illumina HiSeq RNAseq dataset (
n = 186) through the University of California Santa Cruz Cancer Browser (
https://xenabrowser.net/heatmap/) and the GEO Illumina Human HT-12 V4.0 Expression BeadChip GSE59357/GPL10558/GDS5627 dataset on the gene expression profile in pancreatic carcinoma cell lines that are resistant or sensitive to dasatinib, a U.S. FDA-approved small-molecule kinase inhibitor for the treatment of pancreatic cancer (
https://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS5627). We also used the AFFY_HG_U133_PLUS_2 dataset GSE17891/GPL570, which originally investigated the pervasive subtypes of PDAC and their different responses to anticancer treatment (
n = 47 samples, 54,675 genes) (
https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE17891&platform=GPL570).
2.2. Drugs and Reagents
PD173074 (Sigma-P2499, HPLC ≥ 96%) and olaparib (AZD2281/KU0059436, #S1060, HPLC ≥ 99.7%) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) and Selleck Chemicals (Antibody International Inc. Jhubei City, Hsinchu County, Taiwan), respectively. Stock solutions (1 mM) of each drug were prepared by dissolution in phosphate-buffered saline (PBS) and stored in a dark room at −20 °C. PBS, dimethyl sulfoxide (DMSO), sulforhodamine B (SRB) reagent, trypsin/ethylenediaminetetraacetic acid, Tris aminomethane (Tris) base, and acetic acid were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Invitrogen (Invitrogen Life Technologies, Carlsbad, CA, USA).
2.3. Cell lines and Culture
Human PDAC cell lines PANC-1 (ATCC® CRL-1469), AsPC-1 (ATCC® CRL-1682), and PANC 0403 (ATCC® CRL-2555) were obtained from American Type Culture Collection (ATCC Manassas, VA, USA), and SUIT-2 (Japanese Collection of Research Bioresources Cell Bank [JCRB]1094) cells were obtained from the National Institute of Biomedical Innovation, Health and Nutrition (JCRB Cell Bank, Japan). The PANC-1 and SUIT-2 cells were cultured in DMEM (Invitrogen Life Technologies, Carlsbad, CA, USA). Culture media were supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (Invitrogen, Life Technologies, Carlsbad, CA, USA). The cells were incubated in a 5% humidified CO2 incubator at 37 °C. The cells were subcultured at 100% confluence every 48–72 h. The vendors identified and authenticated the cell lines on the basis of karyotype and short tandem repeat analyses, and our team regularly checked the cells to confirm that they were free from mycoplasma contamination. The PDAC cells were treated with indicated concentrations of olaparib and/or PD173074.
2.4. Sulforhodamine B Cytotoxicity Assay
The PANC-1 or SUIT-2 cells were seeded at a density of 3 × 103 cells/well in 96 well plates in triplicate and were cultivated for 24 h. Then, the cells were treated with olaparib and/or PD173074 for 48 h, fixed with 10% trichloroacetic acid, washed carefully with double-distilled water, and stained using a 0.4% 0.4: 1 (w/v) SRB/acetic acid solution. After the unbound dye was removed by carefully washing three times with 1% acetic acid before air-drying the plates, the bound SRB dye was solubilized in 10 mM Tris base, and absorbance was read at 570 nm by using a Molecular Devices Spectramax M3 multimode microplate reader (Molecular Devices LLC., San Jose, CA, USA). The absorbance was positively correlated with cell number over a wide range of values.
2.5. Western Blot Analysis
Protein samples (20 μg) were separated using a 10% sodium dodecyl sulfate-polyacrylamide electrophoresis gel and transferred to polyvinylidene fluoride (PVDF) membranes by using a Bio-Rad Mini-Protein electro-transfer system (Bio-Rad Laboratories, Inc., CA, USA). Next, the PVDF membranes were blocked in 5% nonfat milk in Tris-buffered saline with Tween 20 for 1 h. After blocking, the membranes were incubated at 4 °C overnight with primary antibodies against FGFR1 (#9740; 1:1000, Cell Signaling Technology Inc., Danvers, MA, USA), PARP (#9532; 1:1000, Cell Signaling Technology), cleaved PARP (#5625; 1:1000, Cell Signaling Technology), caspase-9 (#9508; 1:1000, Cell Signaling Technology), caspase-3 (#9668; 1:1000, Cell Signaling Technology), Bcl-xL (#2764; 1:1000, Cell Signaling Technology), and β-actin (sc-69879; 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA). All the antibodies used are listed in
Supplementary Table S1. The membranes were then incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h and washed carefully with PBS three times. Next, protein band detection was performed using the enhanced chemiluminescence detection system (Thermo Fisher Scientific Inc., Waltham, MA, USA). The protein bands were quantified using ImageJ (
https://imagej.nih.gov/ij/).
2.6. Tumorsphere Formation Assay
The PANC-1 or SUIT-2 cells pretreated with indicated concentrations of olaparib and/or PD173074 for 24 h were seeded at a density of 5 × 104 cells/well in 6 well nonadherent plates (Corning Inc., Corning, NY, USA) in DMEM supplemented with B27 supplement (Invitrogen, Carlsbad, CA, USA), 20 ng/mL basic fibroblast growth factor (Invitrogen, Carlsbad, CA, USA), and 20 ng/mL epidermal growth factor (Millipore, Bedford, MA, USA). The cells were cultured for 2 weeks, and tumorspheres formed were counted using an inverted phase-contrast microscope.
2.7. Colony Formation Assay
The colony formation capability of the cells was evaluated as previously described [
14]. Briefly, the PANC-1 or SUIT-2 cells pretreated with indicated concentrations of olaparib and/or PD173074 for 24 h were seeded at a density of 1 × 10
3 cells/well in 6 well plates and cultured in a 5% humidified CO
2 incubator at 37 °C for 15 days. Thereafter, the colonies formed (>50 cells/colony) were stained with crystal violet dye, photographed, and counted.
2.8. Immunofluorescence Staining
The PANC-1 or SUIT-2 cells were plated at a density of 2 × 104 cells/well in 6 well chamber slides (Nunc™, Thermo Fisher Scientific) for 24 h. For γ-H2AX staining or RAD51 foci quantification to assess DNA damage repair, the seeded cells were incubated with indicated concentrations of olaparib and/or PD173074 for 48 h and fixed with 4% paraformaldehyde at room temperature for 20 min. The cells were then washed with PBS, permeabilized using 0.1% Triton X-100 in 0.01 M PBS (pH 7.4) for 5 min, blocked in 0.2% bovine serum albumin for 1 h, air dried, and rehydrated in PBS. Next, the cells were incubated with a rabbit polyclonal antibody against histone H2AX (#07-627, Merck KGaA, Darmstadt, Germany) or Rad51 (#07-1782, Merck KGaA, Darmstadt, Germany) diluted to 1:500 in PBS containing 3% normal goat serum at room temperature for 2 h, washed three times in PBS for 10 min each, followed by incubation with an anti-rabbit IgG fluorescein isothiocyanate-conjugated secondary antibody (Jackson ImmunoResearch Inc., West Grove, PA, USA) diluted 1:500 in PBS for 1 h at room temperature. The cells were washed in PBS and mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining. Cell imaging was performed using a Zeiss Axiophot fluorescence microscope (Carl Zeiss Microscopy LLC, Thornwood, NY, USA).
2.9. Tumor Xenograft Studies
Female BALB/c mice (4–6 weeks old,
n = 40, median weight = 12.7 ± 2.1 g) were purchased from BioLASCO (BioLASCO Taiwan Co. Ltd., Taipei, Taiwan) and maintained under specific pathogen-free condition with free access to rodent chow and water. The mice were subcutaneously inoculated with 5 × 10
4 PANC-1 tumorsphere cells suspended in 100 μL of serum-free medium. The mice were randomly divided into two treatment regime groups, namely early start (
n = 20; treatment started 72 h after inoculation with PANC-1 cells) and late start (
n = 20; treatment initiated 3 weeks after inoculation with PANC-1 cells) groups. Each group was randomly divided into four treatment subgroups, namely vehicle-treated control, olaparib-treated, PD173074-treated, or combination-treated (olaparib+PD173074) subgroups (
n = 5 in each subgroup). Randomization was conducted by blindly picking numbers from a dish. The mice were treated with 50 mg/kg olaparib and/or 20 mg/kg PD173074 by intraperitoneal injection daily for 5 consecutive days per week until the last day of Week 9, regardless of the treatment group. The mice in the control group were injected with PBS. Tumor volume (mm
3) was measured weekly. The formula used for calculating volume was volume = (L × W
2)/2, where L and W represent the longest and shortest diameters, respectively. The mice were humanely sacrificed at the end of the experimental period, and tumors were resected and weighed. All animal experiments were approved by the Institutional Animal Care and Use Committee of National Defense Medical Center (IACUC-19-101), and all experiments complied with guidelines provided in The National Academies of Science, Engineering and Medicine Guide for the Care and Use of Laboratory Animals [
15].
2.10. Immunocytochemical Staining
Sections from tumor samples resected from the PD173074 and/or olaparib-treated tumor-bearing BALB/c mice were stained using primary antibodies against FGFR1 (#9740; 1:400, Cell Signaling Technology Inc., Danvers, MA, USA), cleaved PARP (#5625; 1:400, Cell Signaling Technology), Ki-67 (#ab15580; 1:500, Abcam, Cambridge, MA, USA), and Bcl-2 (#15071; 1:400, Cell Signaling Technology). Furthermore, immunohistochemical staining was performed according to the manufacturer’s instructions. Briefly, after washing coverslips with PBS (pH 7.4), they were incubated in 3% H2O2 for 10 min and then treated with appropriate primary antibodies at 37 °C in a humid chamber for 60 min. Next, the coverslips were stained with the biotinylated HRP-conjugated streptavidin system (#N100, Thermo Fisher Scientific Inc., Bartlesville, OK, USA) before imaging under microscope.
2.11. Statistical Analysis
All data represent the mean ± standard deviation of experiments performed at least three independent times in triplicates. A two-sided Student’s t-test was used for comparing between two groups, whereas one-way analysis of variance and Tukey’s post hoc test were used for multiple comparisons. Cumulative survival rates were calculated using the Kaplan–Meier method, and the significance of differences in the survival rate was analyzed using the log-rank test. Statistical analyses were performed using GraphPad Prism Version 7 for Windows (GraphPad Software, Inc., San Diego, CA, USA). A p-value of <0.05 was considered statistically significant.
4. Discussion
Despite improved understanding of PDAC biology alongside numerous diagnostic and therapeutic advancements made in the management of patients with PDAC in the last two decades, the therapeutic success and prognosis of PDAC remain dismal. This necessitates the discovery of new molecular targets and/or the development of novel therapeutic strategies with evidence-based efficacy against PDAC aggression and molecular events implicated in tumor initiation, distant dissemination, innate or acquired resistance to therapy, and therapy failure in patients with PDAC [
3,
4,
19,
20,
21].
Corroborating the findings of our previous study on the role of aberrant FGFR1 signaling in driving PDAC oncogenicity and stem cell-like phenotype, the therapeutic targetability of FGFR1, and the anti-PDAC efficacy of PD173074, a selective FGFR1 inhibitor [
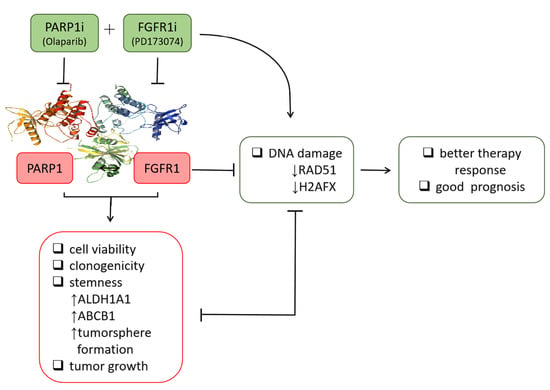
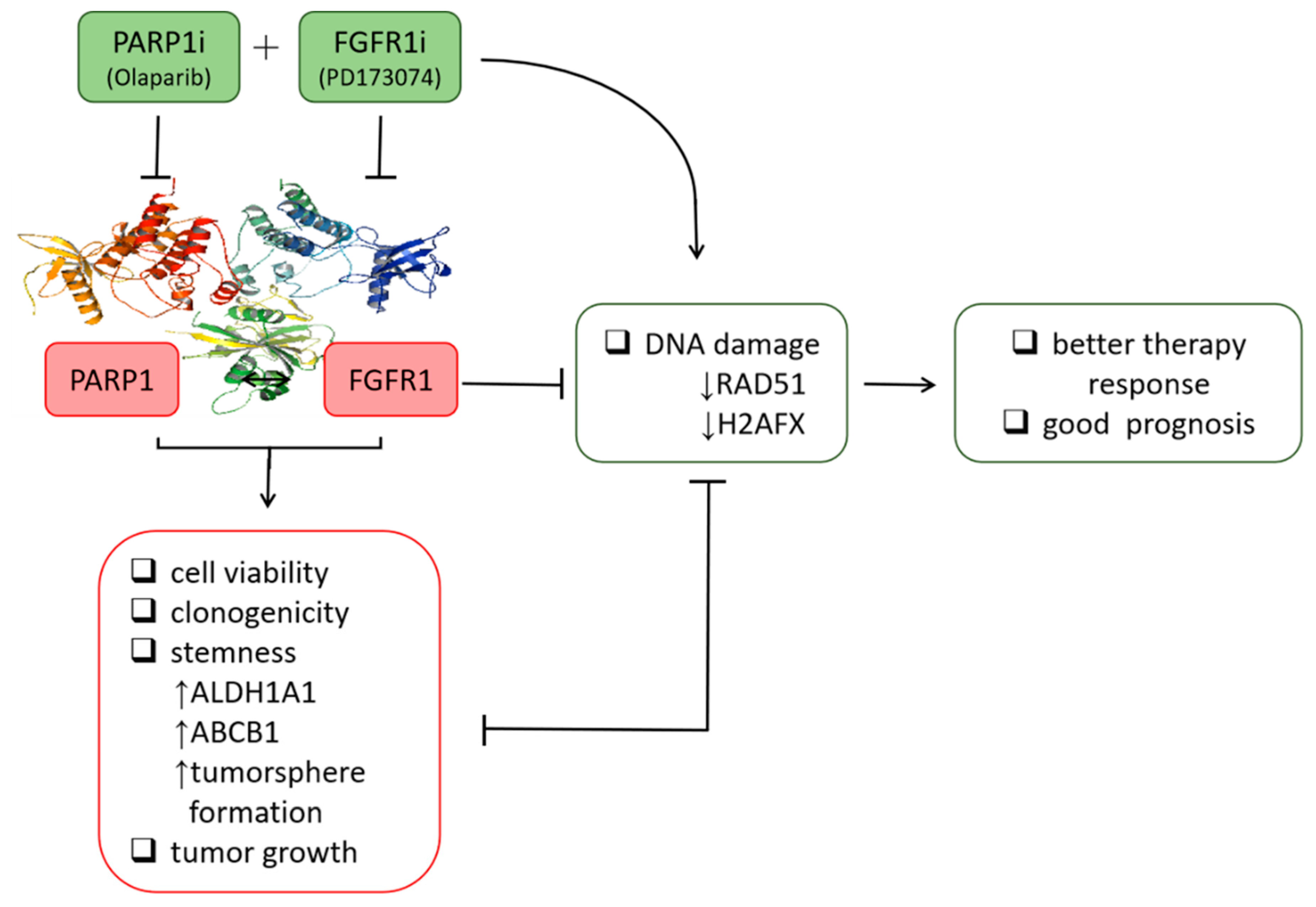
13], the present study provided better mechanistic insight into the therapeutic exploitability of aberrantly expressed FGFR1 in PDAC. First, we demonstrated that enhanced FGFR1 and PARP1 expression characterized FGFR1 inhibitor-resistant pancreatic cells at mRNA and protein levels. Secondly, we showed that the observed aberrant FGFR1 and PARP1 expression was associated with cancer stem cell-like phenotype, regulated DNA repair, and modulated the response to therapy in PDAC cells. Thirdly, our data indicated that FGFR1 and PARP1 co-occurred and directly interacted with each other at the expense of RAD51/H2AFX signaling and had adverse prognostic implications. Fourthly, we provided preclinical evidence that olaparib-induced pharmacological inhibition of PARP1 synergistically enhanced the therapeutic effect of the FGFR1 inhibitor PD173074. Fifthly, we demonstrated that olaparib alone or synergistically with PD173074 suppressed the oncogenic cancer stem cell-like phenotype of PDAC cells through the apoptosis-related impairment of DNA repair. Finally, our results showed that PDAC cancer stem cells were more sensitive to the FGFR1 inhibitor PD173074 in the absence of PARP1 in vivo. These findings improve our current understanding of PDAC biology and may help inform future therapeutic decision-making in the management of patients with PDAC, considering the present clinical challenge of therapy failure and poor prognosis in PDAC cohorts.
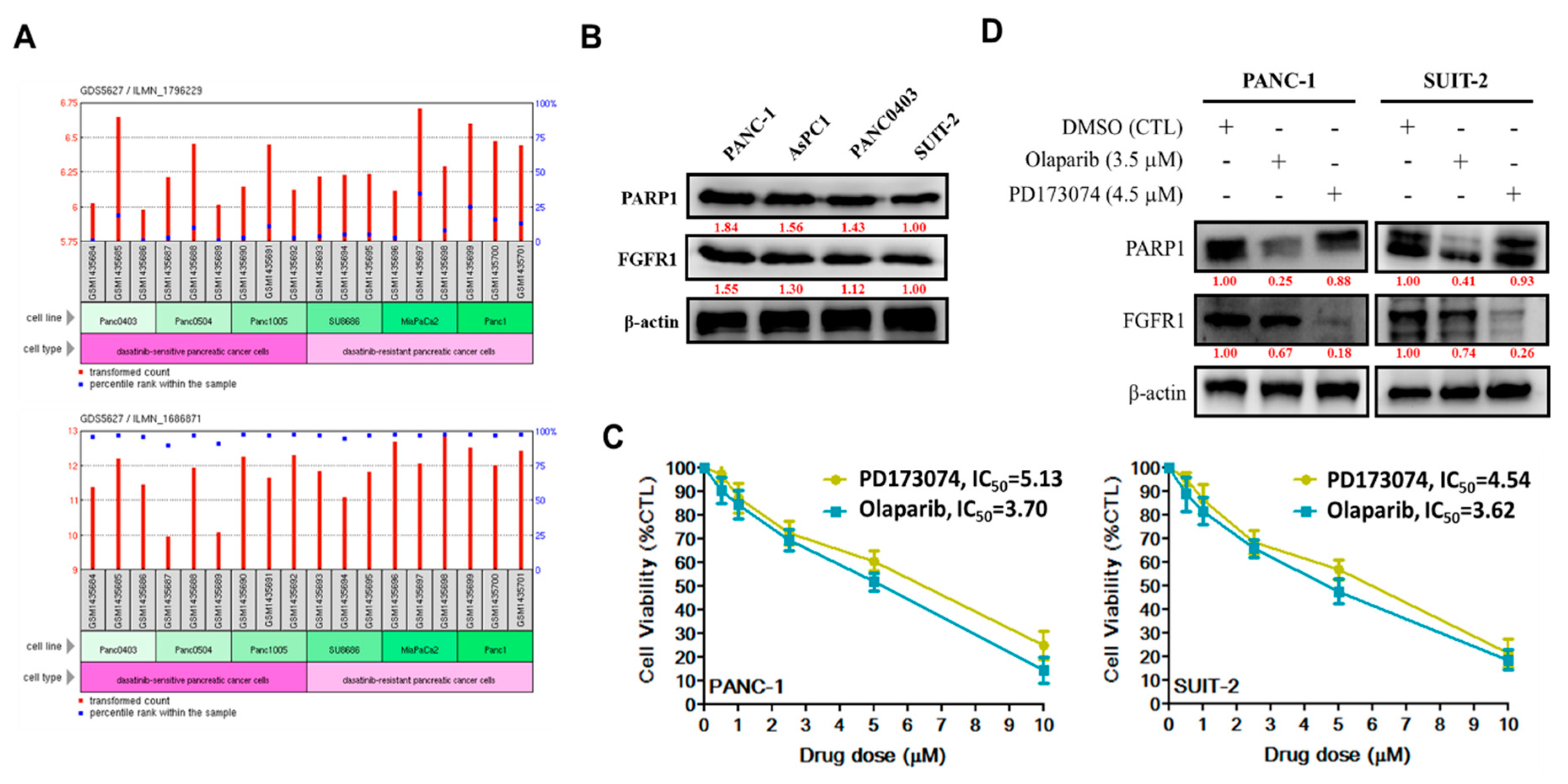
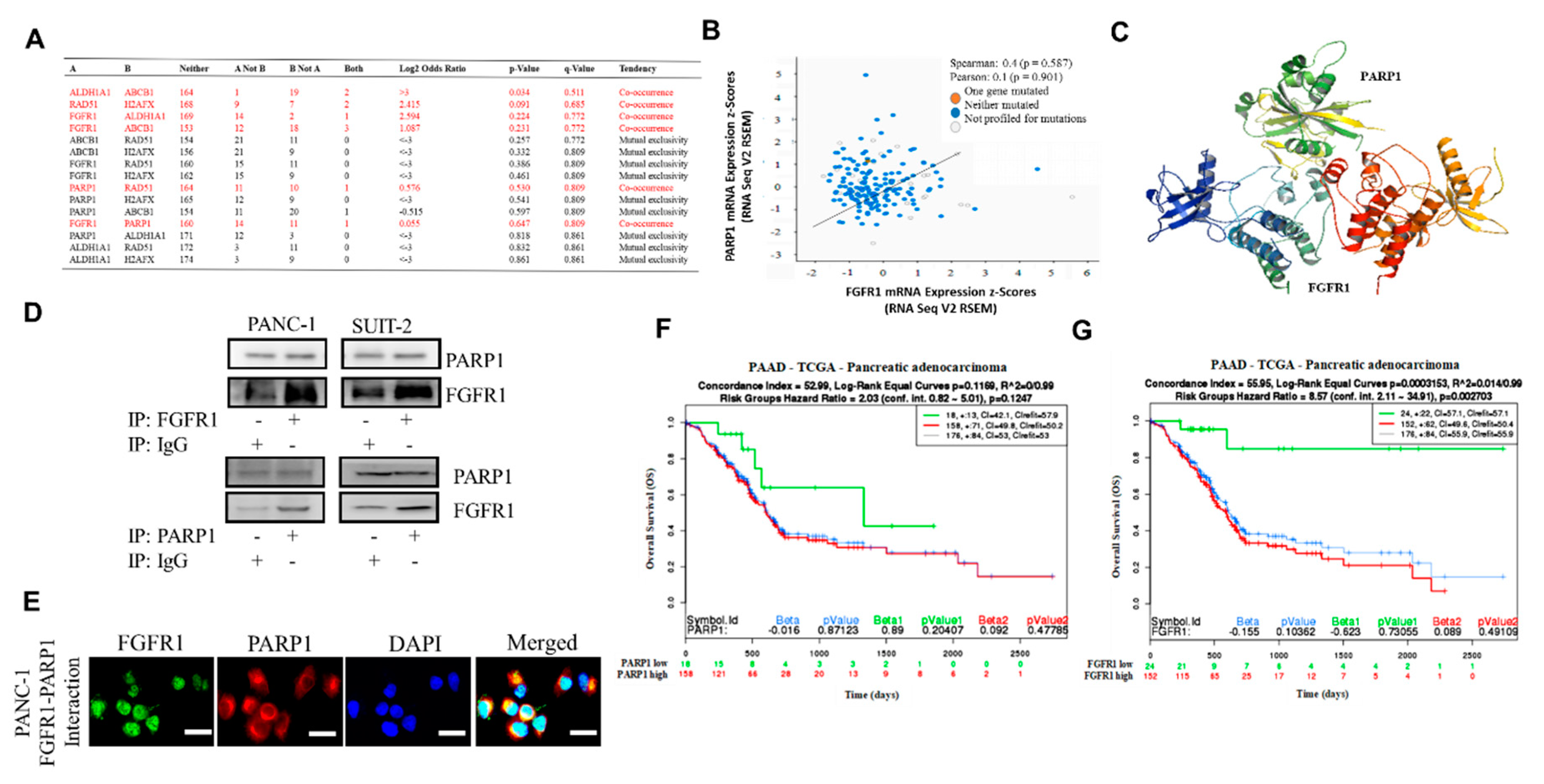
To the best of our knowledge, the present study is the first to demonstrate that resistance to the FGFR1 inhibitor, dasatinib, is partly associated with enhanced FGFR1 and PARP1 expression at mRNA and protein levels (
Figure 1). This finding was corroborated by previous findings indicating that FGFR1 amplification was significantly associated with overexpression of FGFR1, drove anchorage-independent cell proliferation, and enhanced resistance to endocrine therapy [
22,
23], as well as mediated resistance to cyclin-dependent kinase 4/6 inhibitors in breast cancer [
24]. Moreover, our findings were corroborated partly by the results of another study, indicating that the inhibition of PARP-1 activity was elicited by induction of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling with activation of caspase-3 and -8, which sensitized TRA-8-resistant PANC-1 and SUIT-2 cells to the apoptotic action of a monoclonal agonist antibody, which specifically targeted death receptor 5 [
25]. Although our present study did not address the role of caspase-8 in the re-sensitization of resistant PANC-1 and SUIT-2 cells to therapy, we believe that though the induction of DNA damage was not necessary for the induction of cell death or apoptosis by death ligands (DLs), DLs elicited DNA damage in surviving cancerous cells. Thus, based on the findings of Yuan et al., we suggest that FGFR1- and PARP-rich pancreatic cancer cells maintain their insensitivity to single-agent FGFRs and evade cell death by suppressing TRAIL-associated apoptotic DNA fragmentation by caspase-activated DNase/DNA fragmentation factor 40 endonuclease, thus suggesting a critical role of DNA damage/repair balance in pancreatic cancer therapy response [
26,
27].
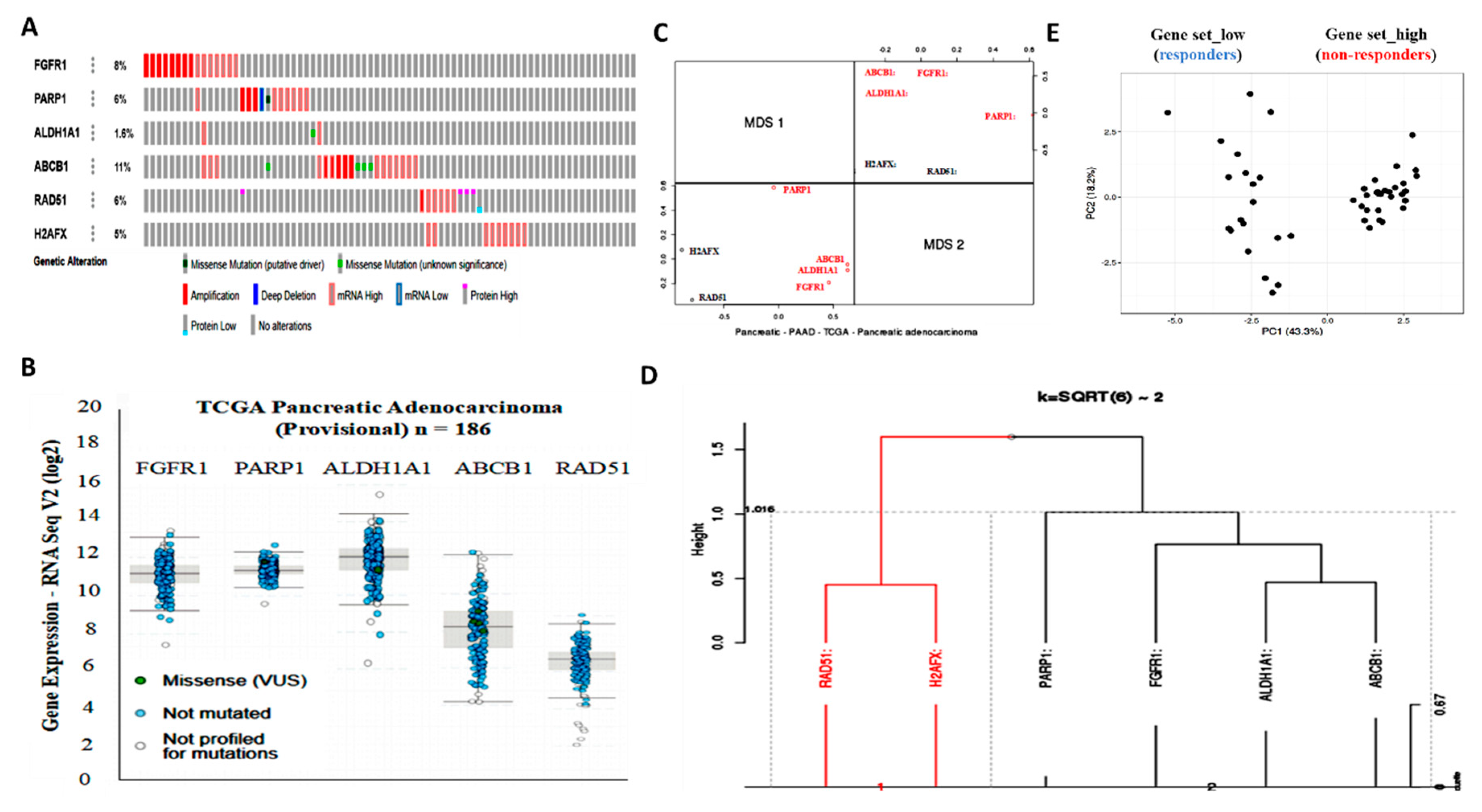
We demonstrated that aberrant FGFR1 and PARP1 expression was associated with cancer stem cell-like phenotype, regulated DNA repair, and modulated response to therapy in PDAC cells. We also reported that the upregulated nested cluster of FGFR1/PARP1/ALDH1A1/ABCB1 was inversely correlated to and diametrically distant from the downregulated cluster of H2AFX/RAD51 (
Figure 2). These findings were consistent with our current understanding of the critical roles of the cancer stem cell marker ALDH1A1 and the multidrug resistance protein ATP binding cassette subfamily B member 1 (ABCB1/MDR1) in the enhancement of tumorigenicity, activation of DNA repair, and resistance to anticancer therapy [
28]. Moreover, our findings are particularly relevant when viewed in the context of recently published work indicating that aberrant FGFR1 expression in the tumor vascular niche was implicated in the transformation of indolent cancer cells into chemoresistant cancer stem cells and that FGFR1+ cells were associated with the engraftment of chemoresistant cancer stem cells [
29]. Similarly, PARP1 has been shown to modulate sensitivity to therapy, and olaparib-based therapy significantly enhanced the sensitivity of pancreatic cancer cells (particularly the p53 mutant cells) and other isogenic cancer cell lines to radiotherapy, by inducing cell cycle arrest in G2, homologous recombination repair inhibition, and persistent DNA damage responses [
30].
The implication of FGFR1, PARP1, ALDH1A1, ABCB1, RAD51, and H2AFX in therapy response was highlighted by our results. The patients with PDAC were stratified into therapy responders and non-responders.
Gene set_high patients exhibiting high FGFR1, PARP1, ALDH1A1, and ABCB1 expression, but low RAD51 and H2AFX expression levels were less sensitive to erlotinib and gemcitabine compared to the
Gene set_low patients who had low FGFR1, PARP1, ALDH1A1, and ABCB1 expression, but high RAD51 and H2AFX expression level (
Figure 2). We posit that the relative non-responsiveness of the
Gene set_high patients may be related to their low mutagenicity, a constitutively active DNA damage response, but impaired replication stress response due to probable suppressed levels of phospho-H2AX and RAD51 at baseline [
31]; this hypothesis was consistent with our results showing that FGFR1 and PARP1 co-occurred and directly interacted with each other at the expense of RAD51/H2AFX signaling and with adverse prognostic implications (
Figure 3). However, a limitation to this current study remains. It has been established that FOLFIRINOX followed by PARP inhibitor (olaparib) was a maintenance therapy for BRCA mutant pancreatic cancer patients. We have yet to determine if FGFR inhibition could result in the downregulation of BRA1/2 and PALB2, since the synthetic lethal interaction between HR deficiency and PARP inhibition has been shown. This issue is currently being investigated in our laboratory to provide further insights into the mechanistic evidence.
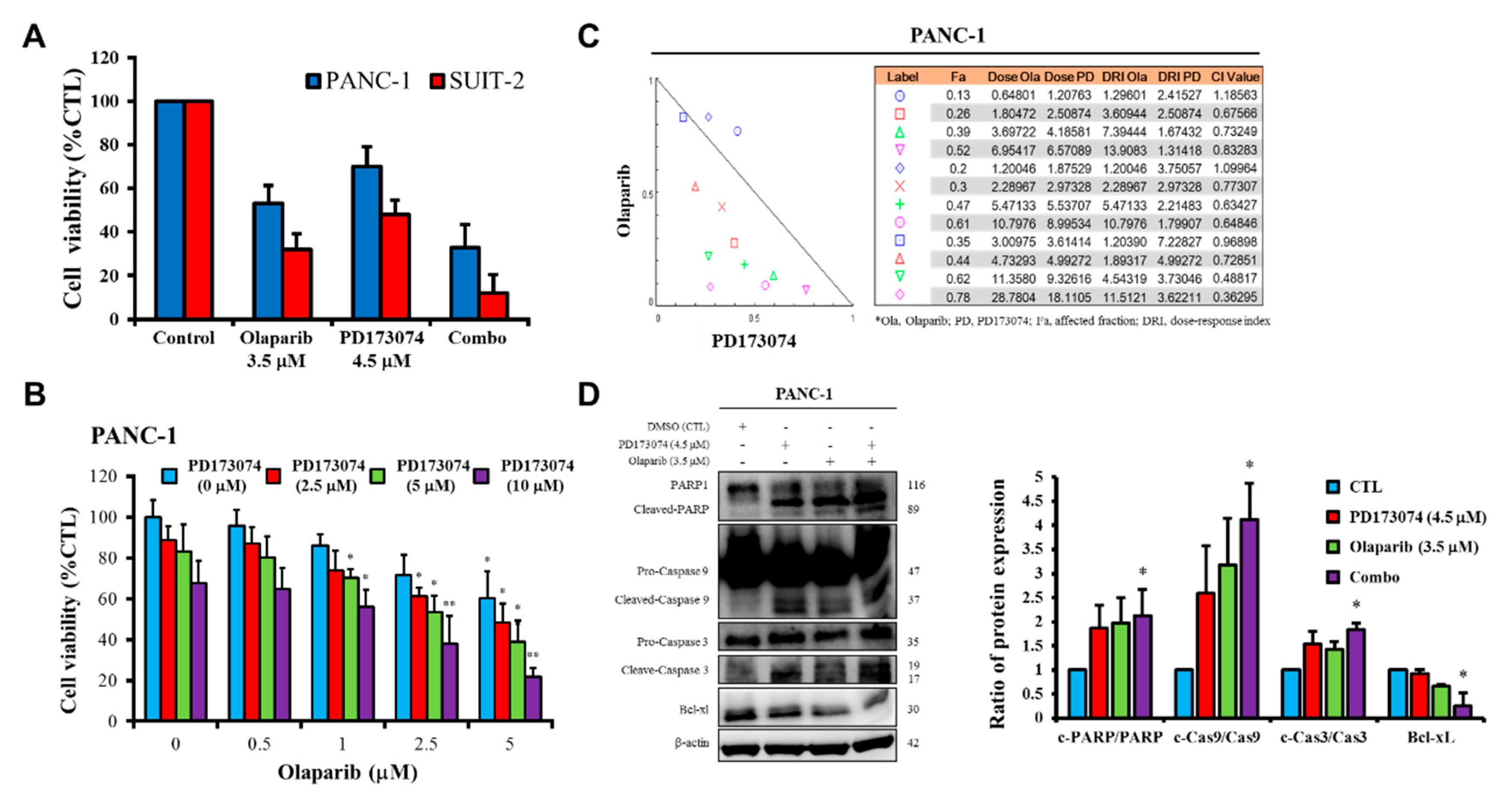
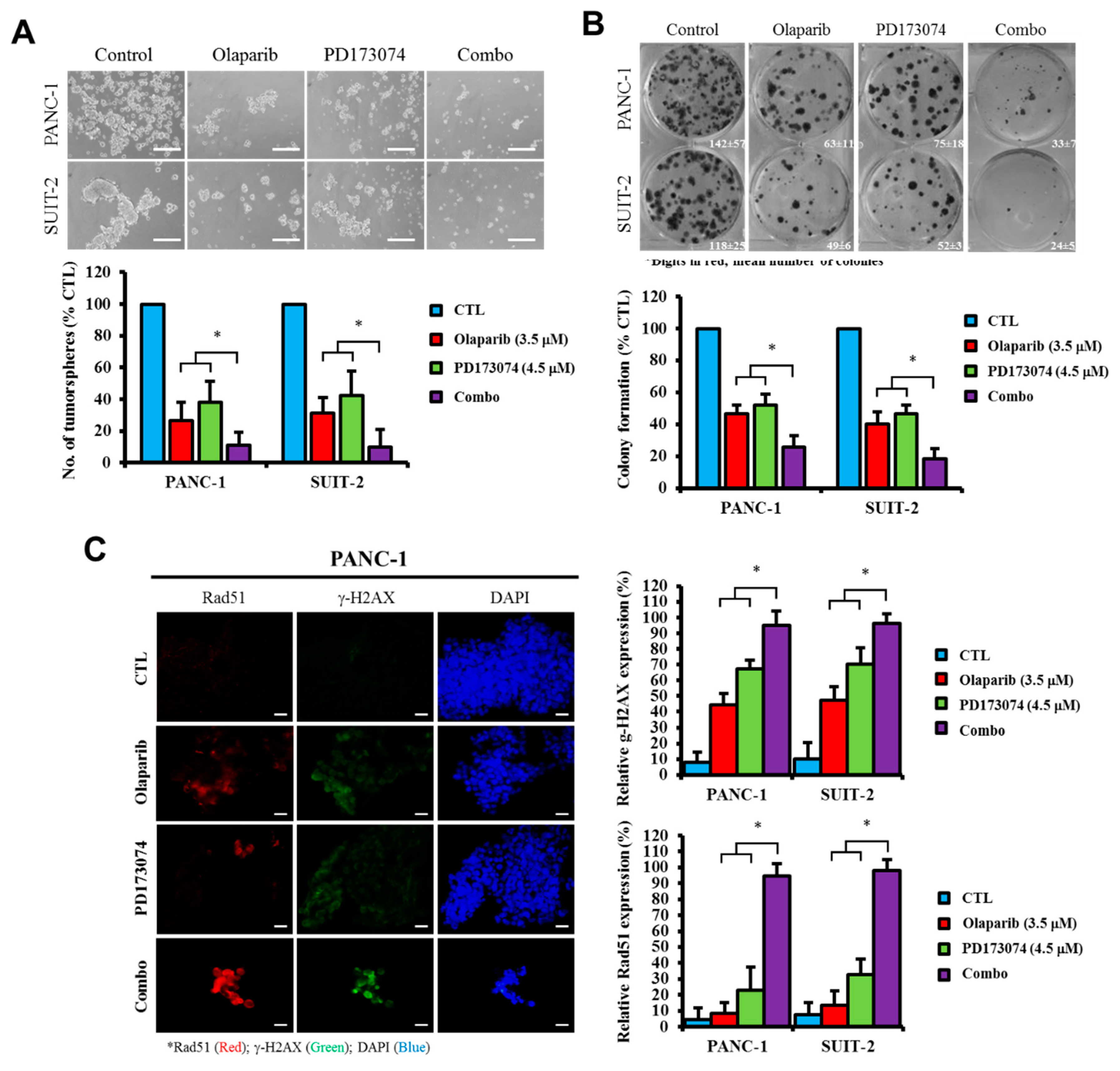
Consistent with the understanding that exposure to PARP inhibitors can enhance the burden of unrepaired DNA double-strand breaks (DSBs) by impeding PARP1 activity and PARP1 trapping onto damaged DNA [
32], we demonstrated that olaparib-induced pharmacological inhibition of PARP1 synergistically enhanced the therapeutic effect of the FGFR1 inhibitor PD173074 and that olaparib alone or synergistically with PD173074 suppressed the oncogenic cancer stem cell-like phenotype of PDAC cells through the apoptosis-related impairment of DNA repair (
Figure 4 and
Figure 5). These results are consistent with recent findings that impairments in DNA repair and accumulation of lethal DNA DSBs induced by tyrosine kinase inhibitors sensitize quiescent and proliferative acute myeloid leukemia stem cells to PARP inhibitors olaparib and BMN673 [
33], indicating synthetic lethality, a biological phenomenon wherein cell death is induced more efficiently by the simultaneous loss of function of multiple molecular targets or genes compared with the loss of function of a single gene [
34]. Similarly, treatment with pan-kinase inhibitors or selective RTKi, namely HS10241, with the PARPi fluzoparib synergistically suppressed tumor growth in multiple cancer types both in vitro and in vivo [
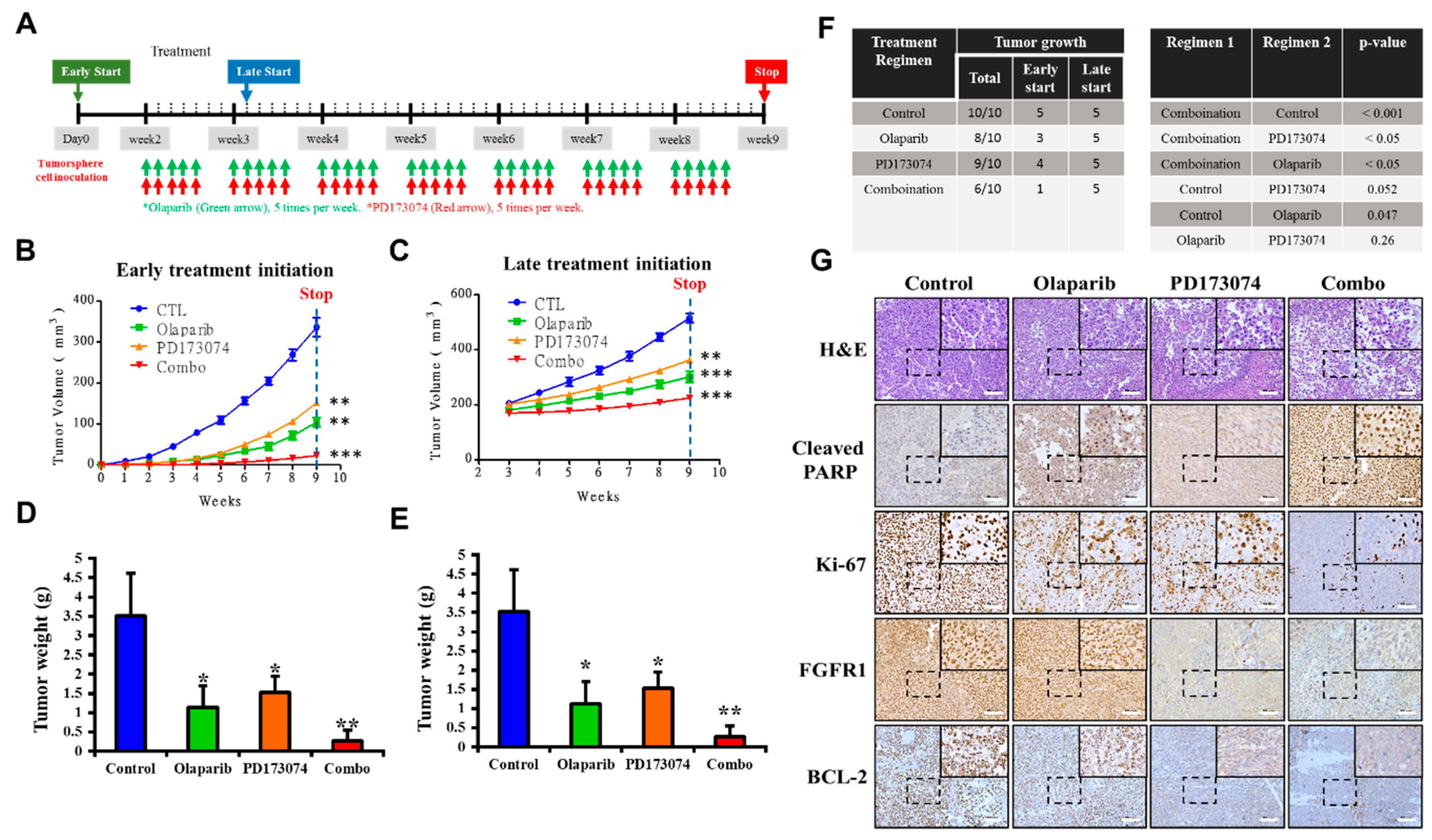
32], which is consistent with our findings indicating that PDAC cancer stem cells were more sensitive to the FGFR1 inhibitor PD173074 in the absence of PARP1, in vivo (
Figure 6).

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






