Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate



Abstract
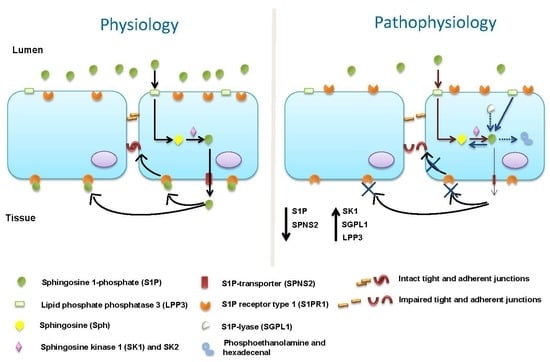
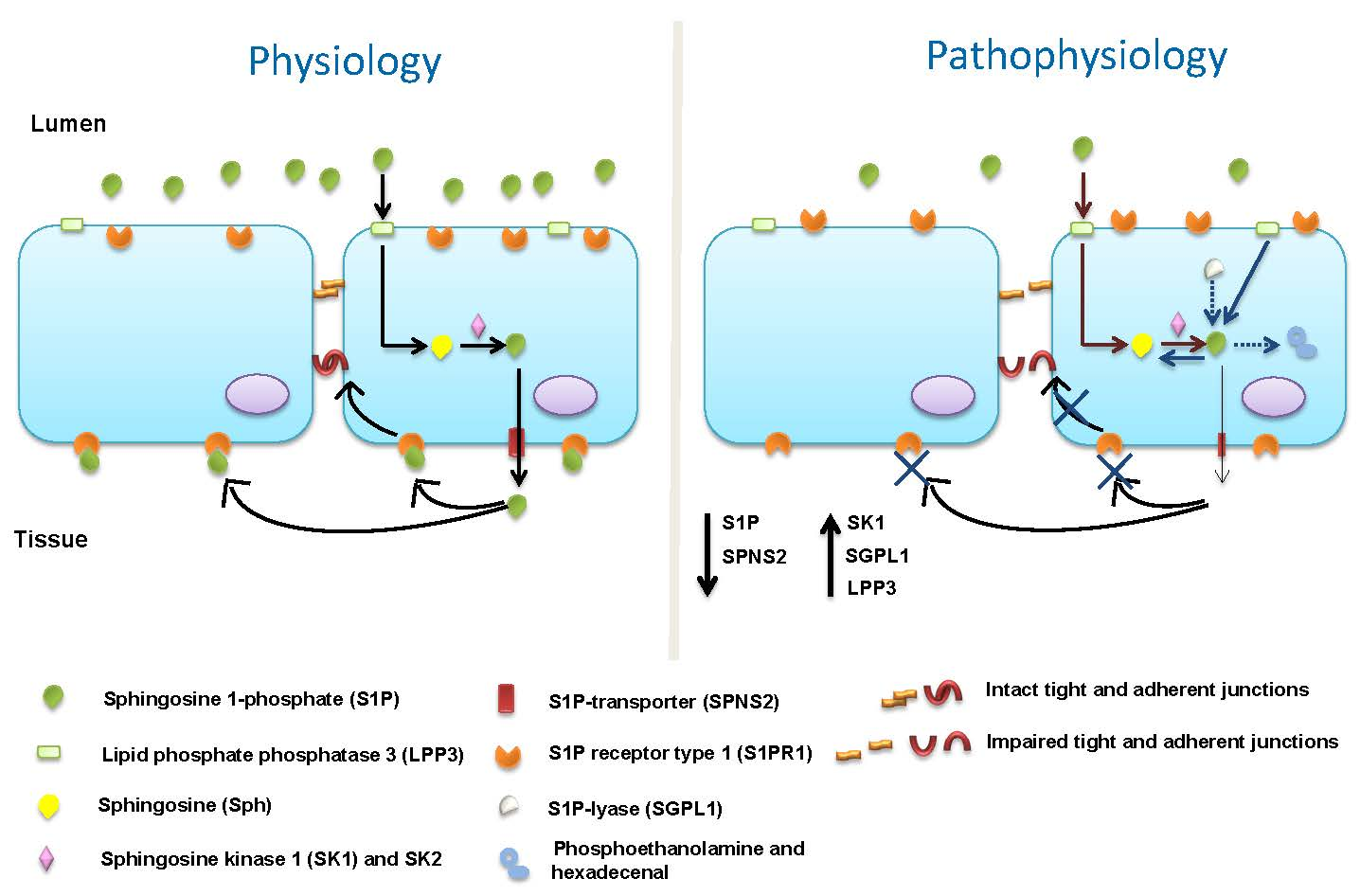
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Isolation of Primary Lung Endothelial Cells
2.3. cDNA Synthesis and Quantitative Polymerase Chain Reaction (qPCR)
2.4. Agarose Gel Electrophoresis
2.5. Flow Cytometry
2.6. Western Blot and Calcium Measurement
2.7. Electric Cell-Substrate Impedance Sensing (ECIS)
2.8. Fluorescence Microscopy
2.9. Measurement of S1P and Sphingosine
2.10. FITC-Dextran Leakage Assay
2.11. Albumin Measurement
2.12. Reagents
2.13. Statistics
3. Results
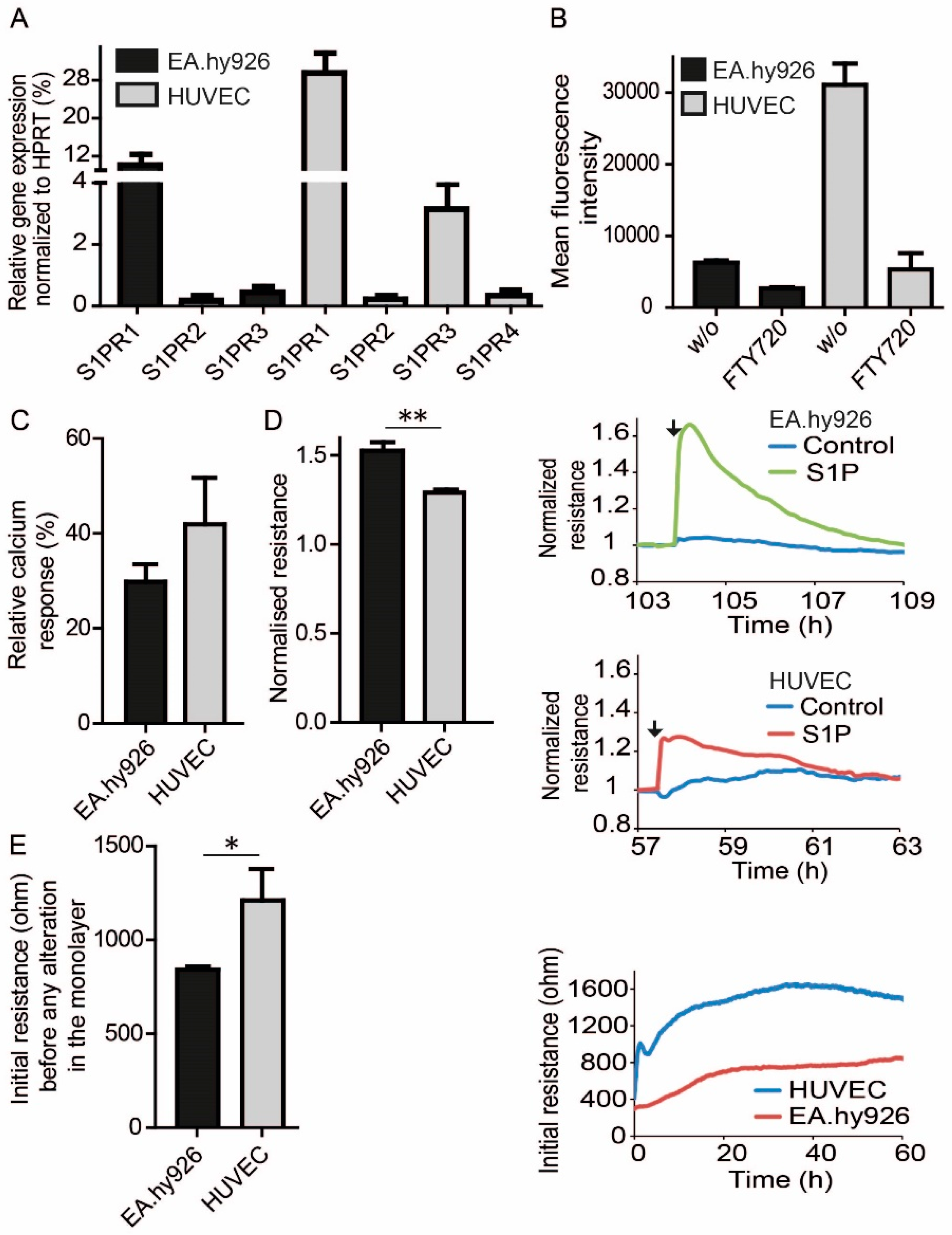
3.1. EC Barrier Stabilizing Function of S1P and S1PR1
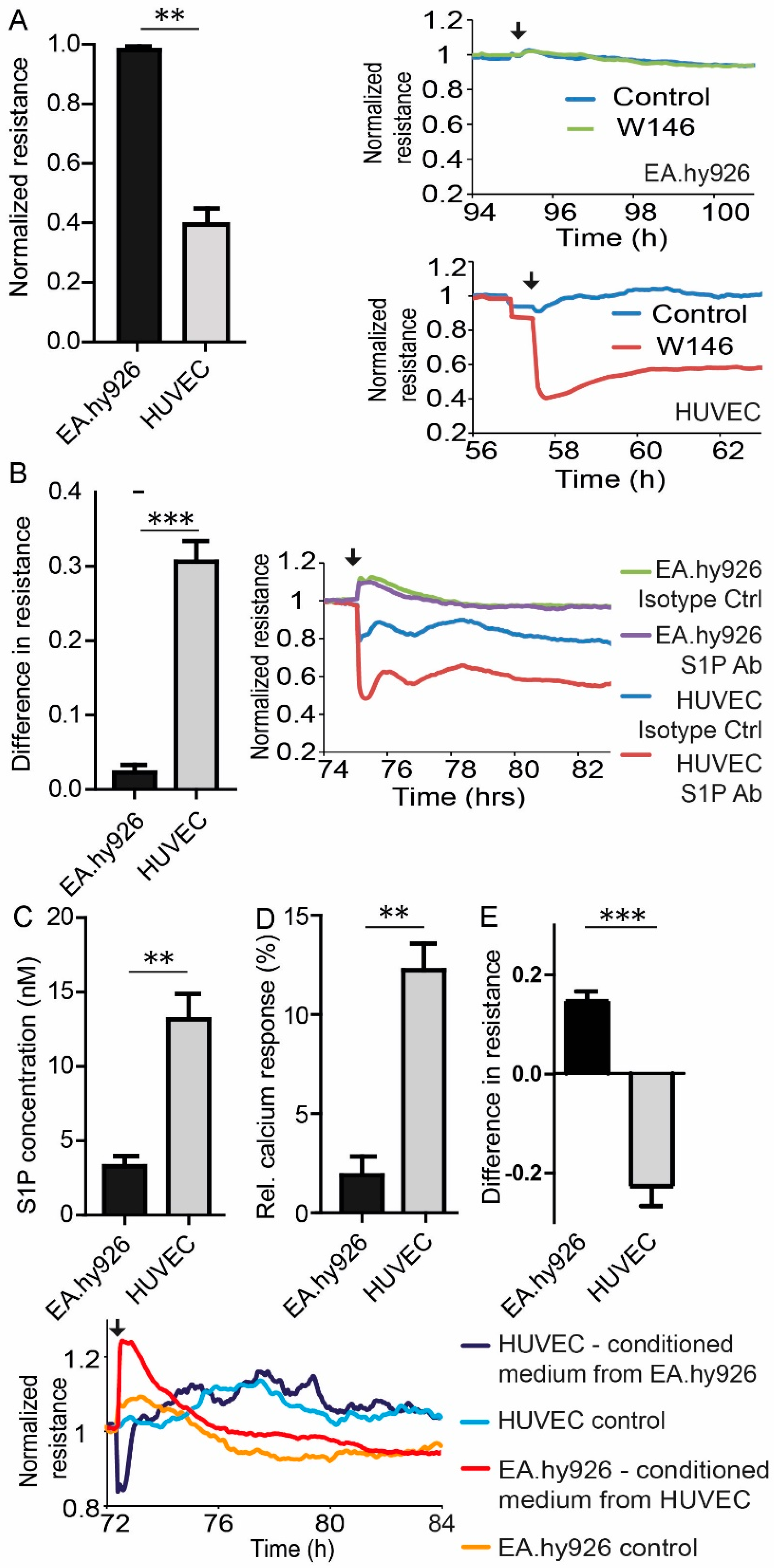
3.2. Endogenous Differences in S1P Signaling between HUVEC and EA.hy926
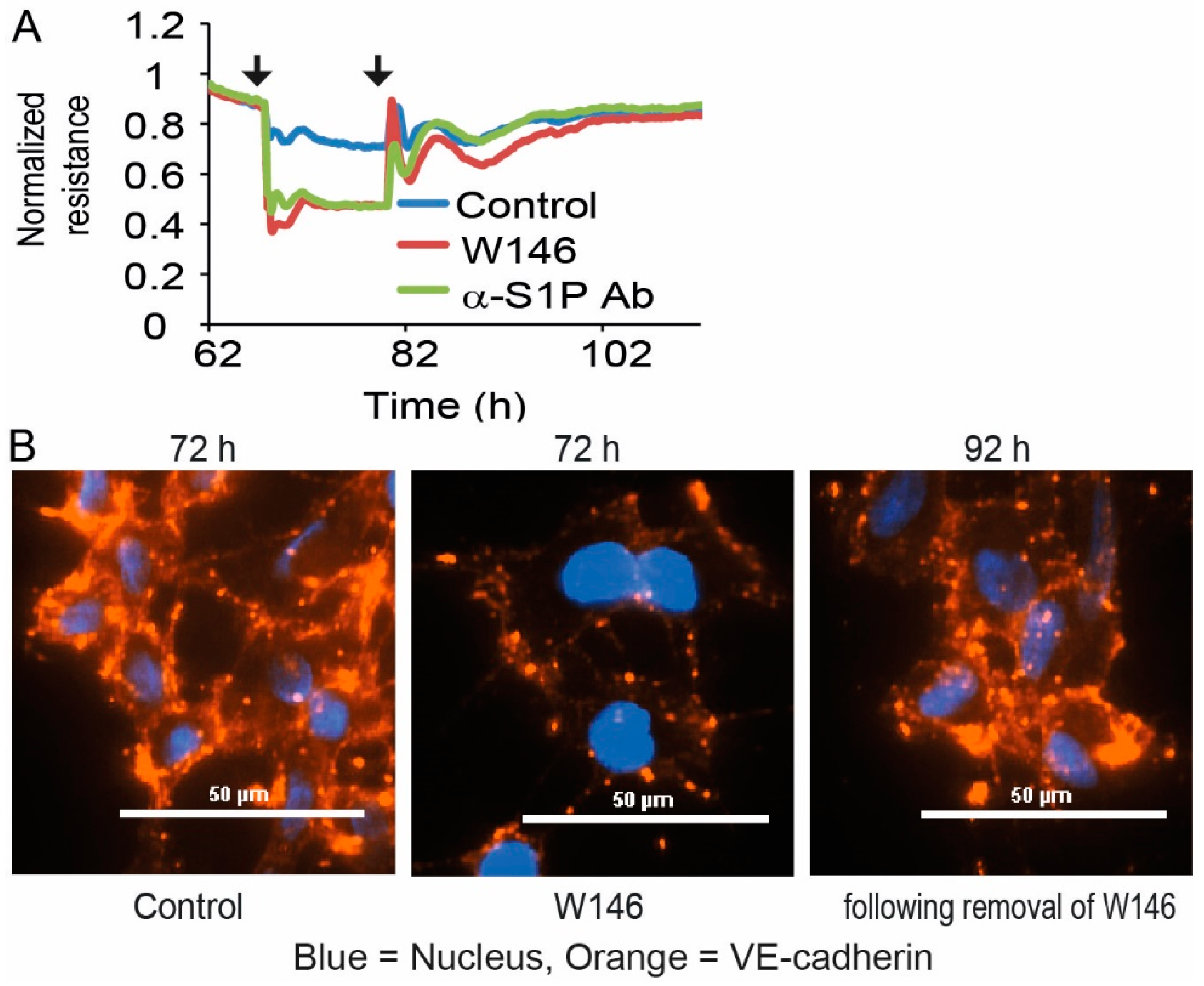
3.3. Reversibility and VE-Cadherin Disturbance of EC Barrier Destabilization by S1PR1 Antagonism and S1P Blocking
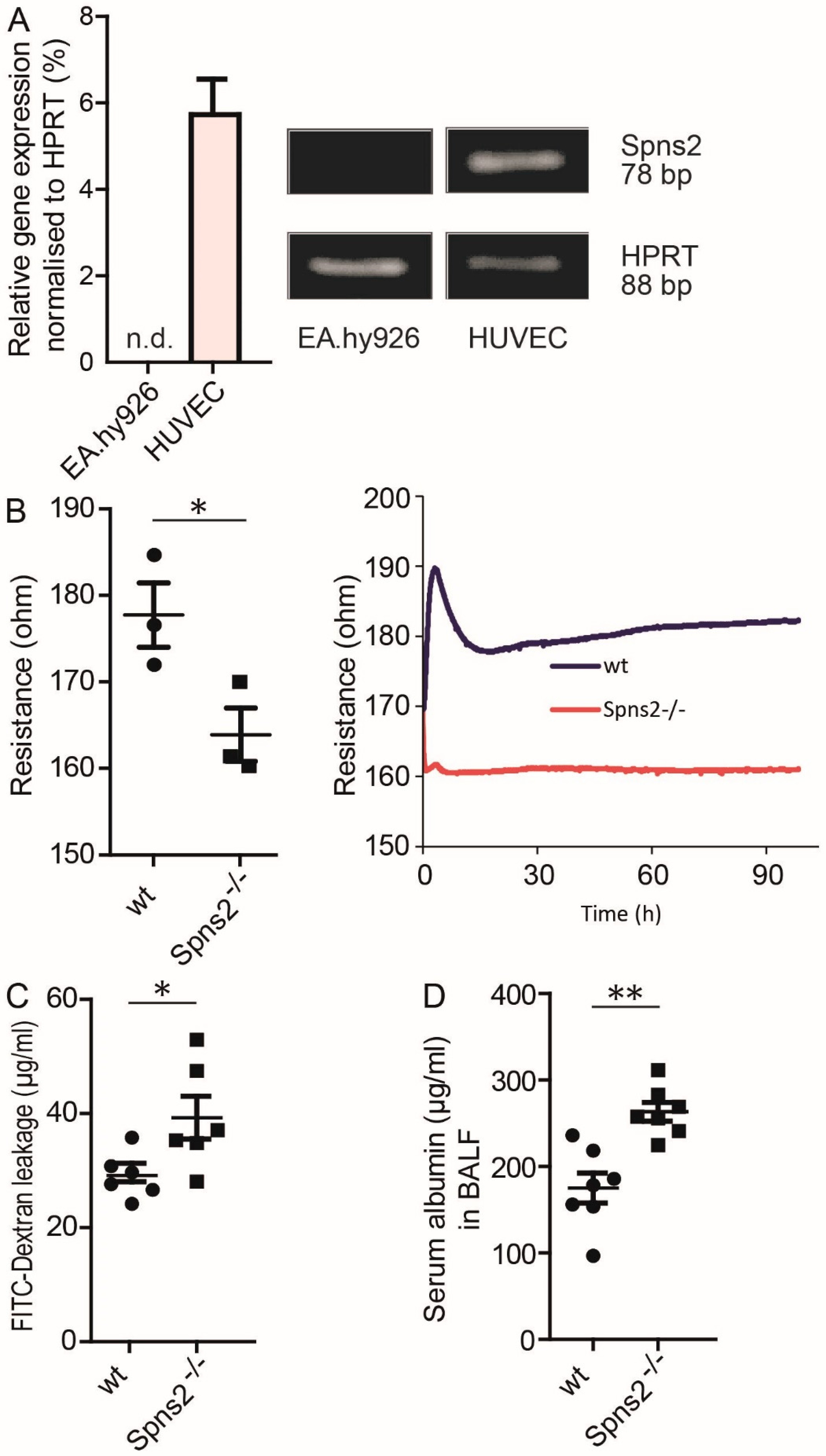
3.4. Role of Spns2 in EC Barrier Maintenance
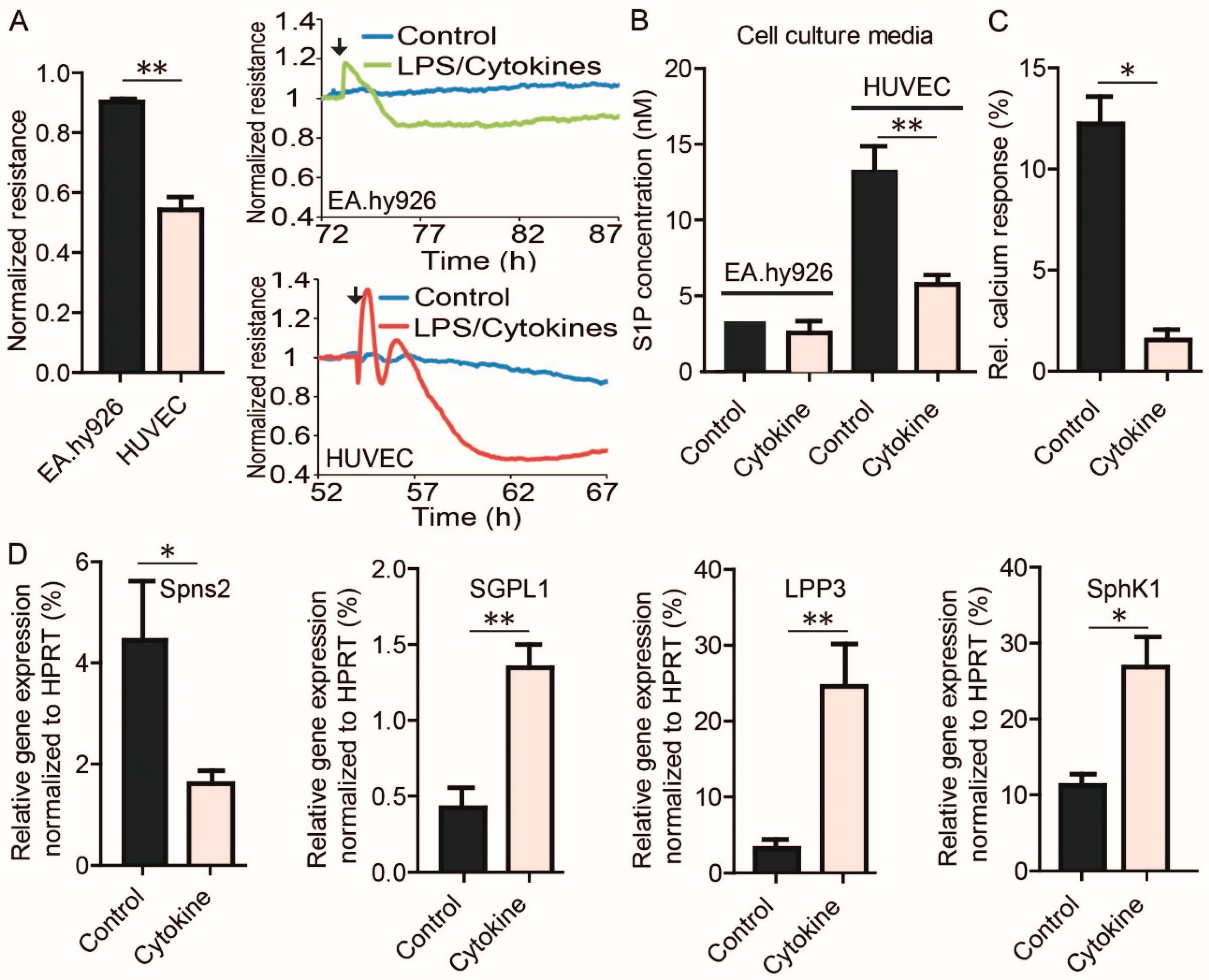
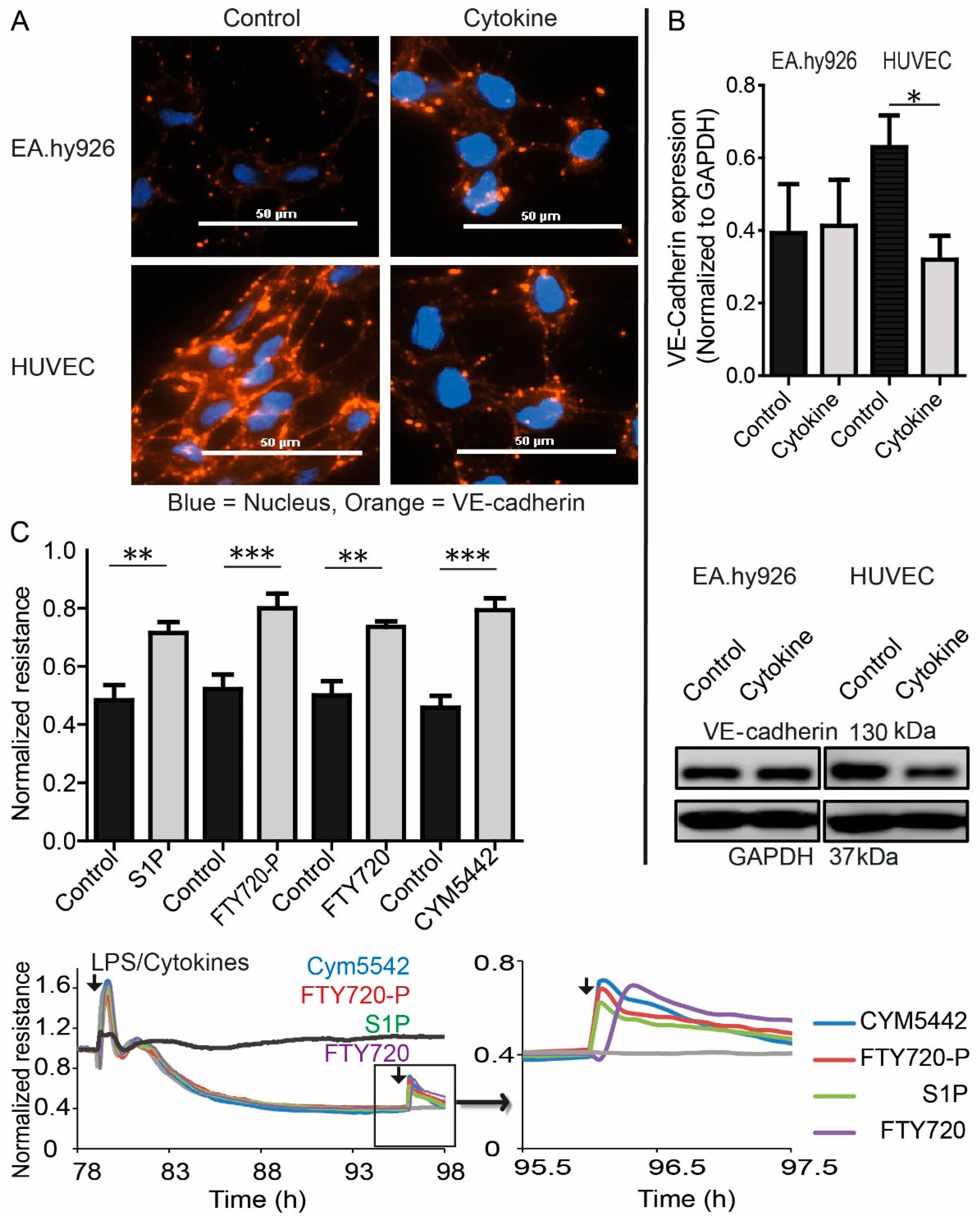
3.5. S1P-Mediated EC Barrier Maintenance Under Inflammatory Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stevens, T.; Garcia, J.G.; Shasby, D.M.; Bhattacharya, J.; Malik, A.B. Mechanisms regulating endothelial cell barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L419–L422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasileva, E.; Citi, S. The role of microtubules in the regulation of epithelial junctions. Tissue Barriers 2018, 6, 1539596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- McVerry, B.J.; Garcia, J.G. Endothelial cell barrier regulation by sphingosine 1-phosphate. J. Cell. Biochem. 2004, 92, 1075–1085. [Google Scholar] [CrossRef]
- Allende, M.L.; Yamashita, T.; Proia, R.L. G-protein-coupled receptor s1p1 acts within endothelial cells to regulate vascular maturation. Blood 2003, 102, 3665–3667. [Google Scholar] [CrossRef]
- Burg, N.; Swendeman, S.; Worgall, S.; Hla, T.; Salmon, J.E. Sphingosine 1-phosphate receptor 1 signaling maintains endothelial cell barrier function and protects against immune complex-induced vascular injury. Arthritis Rheumatol. 2018, 70, 1879–1889. [Google Scholar] [CrossRef]
- Camerer, E.; Regard, J.B.; Cornelissen, I.; Srinivasan, Y.; Duong, D.N.; Palmer, D.; Pham, T.H.; Wong, J.S.; Pappu, R.; Coughlin, S.R. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Invest. 2009, 119, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Garcia Ponce, A.; Vadillo, E.; Pelayo, R.; Rossaint, J.; Zarbock, A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. 2017, 74, 1985–1997. [Google Scholar] [CrossRef]
- Coldewey, S.M.; Benetti, E.; Collino, M.; Pfeilschifter, J.; Sponholz, C.; Bauer, M.; Huwiler, A.; Thiemermann, C. Elevation of serum sphingosine-1-phosphate attenuates impaired cardiac function in experimental sepsis. Sci. Rep. 2016, 6, 27594. [Google Scholar] [CrossRef] [Green Version]
- Frej, C.; Linder, A.; Happonen, K.E.; Taylor, F.B.; Lupu, F.; Dahlback, B. Sphingosine 1-phosphate and its carrier apolipoprotein m in human sepsis and in Escherichia coli sepsis in baboons. J. Cell. Mol. Med. 2016, 20, 1170–1181. [Google Scholar] [CrossRef]
- Gomes, L.; Fernando, S.; Fernando, R.H.; Wickramasinghe, N.; Shyamali, N.L.; Ogg, G.S.; Malavige, G.N. Sphingosine 1-phosphate in acute dengue infection. PLoS ONE 2014, 9, e113394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.S.; Nierhaus, A.; Holzmann, M.; Mudersbach, E.; Bauer, A.; Robbe, L.; Zahrte, C.; Geffken, M.; Peine, S.; Schwedhelm, E.; et al. Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Crit. Care 2015, 19, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Graler, M.H.; Goetzl, E.J. The immunosuppressant fty720 down-regulates sphingosine 1-phosphate g-protein-coupled receptors. FASEB J. 2004, 18, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, A.; Breart, B.; Ramos-Perez, W.D.; Pitt, L.A.; Gobert, M.; Sunkara, M.; Lafaille, J.J.; Morris, A.J.; Schwab, S.R. The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2012, 2, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sensken, S.C.; Staubert, C.; Keul, P.; Levkau, B.; Schoneberg, T.; Graler, M.H. Selective activation of g alpha i mediated signalling of s1p3 by fty720-phosphate. Cell. Signal. 2008, 20, 1125–1133. [Google Scholar] [CrossRef]
- Bode, C.; Graler, M.H. Quantification of sphingosine-1-phosphate and related sphingolipids by liquid chromatography coupled to tandem mass spectrometry. Methods Mol. Biol. 2012, 874, 33–44. [Google Scholar]
- Gazit, S.L.; Mariko, B.; Therond, P.; Decouture, B.; Xiong, Y.; Couty, L.; Bonnin, P.; Baudrie, V.; Le Gall, S.M.; Dizier, B.; et al. Platelet and erythrocyte sources of s1p are redundant for vascular development and homeostasis, but both rendered essential after plasma s1p depletion in anaphylactic shock. Circ. Res. 2016, 119, e110–e126. [Google Scholar] [CrossRef]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest. 2012, 122, 1416–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, A.; Nishi, T.; Hisano, Y.; Fukui, H.; Yamaguchi, A.; Mochizuki, N. The sphingolipid transporter Spns2 functions in migration of zebrafish myocardial precursors. Science 2009, 323, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahashi, M.; Yamada, A.; Katsuta, E.; Aoyagi, T.; Huang, W.C.; Terracina, K.P.; Hait, N.C.; Allegood, J.C.; Tsuchida, J.; Yuza, K.; et al. Targeting the sphk1/s1p/s1pr1 axis that links obesity, chronic inflammation, and breast cancer metastasis. Cancer Res. 2018, 78, 1713–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettorazzi, S.; Bode, C.; Dejager, L.; Frappart, L.; Shelest, E.; Klassen, C.; Tasdogan, A.; Reichardt, H.M.; Libert, C.; Schneider, M.; et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of sphk1. Nat. Commun. 2015, 6, 7796. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Wang, H.; Zhao, J.; Sun, J.; Zhang, T.; Zuo, L.; Zhu, W.; Gong, J.; Li, Y.; Gu, L.; et al. Sew2871 protects from experimental colitis through reduced epithelial cell apoptosis and improved barrier function in interleukin-10 gene-deficient mice. Immunol. Res. 2015, 61, 303–311. [Google Scholar] [CrossRef]
- Flemming, S.; Burkard, N.; Meir, M.; Schick, M.A.; Germer, C.T.; Schlegel, N. Sphingosine-1-phosphate receptor-1 agonist sew2871 causes severe cardiac side effects and does not improve microvascular barrier breakdown in sepsis. Shock 2018, 49, 71–81. [Google Scholar] [CrossRef]
- Hemdan, N.Y.; Weigel, C.; Reimann, C.M.; Graler, M.H. Modulating sphingosine 1-phosphate signaling with dop or fty720 alleviates vascular and immune defects in mouse sepsis. Eur. J. Immunol. 2016, 46, 2767–2777. [Google Scholar] [CrossRef]
- Fleischmann, R. Novel small-molecular therapeutics for rheumatoid arthritis. Curr. Opin. Rheumatol. 2012, 24, 335–341. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | Probe (5′-56-FAM; 3′-36-TAM) |
|---|---|---|---|
| HPRT | agcctaagatgagagttc | cacagaactagaacattgata | atctggagtcctattgacatcgcc |
| S1PR1 | agcactatatcctcttctg | tgaccaaggagtagattc | tcttcactctgcttctgctctcc |
| S1PR2 | catcgtcatcctctgttg | agtggaacttgctgtttc | ccttctggtgctcattgcgg |
| S1PR3 | ccaagcagaagtaaatcaag | catggagacgatcagttg | agcagcaacaatagcagccac |
| S1PR4 | gcttctgtgtgattctgg | ccatgatcgaacttcaatg | cctctctgggcctcagtagg |
| S1PR5 | ggaacaatgatggagatt | ggcattgtccttgataac | attccactcttacactcaattcctgag |
| SK1 | ggcagcttccttgaacca | gcaggttcatgggtgaca | ctatgagcaggtcaccaatgaagacctcct |
| SK2 | ccgacggcctctcagt | cctggccctgggtctta | acagtgagacctgactccttgctcctacc |
| SGPL1 | cctagcacagaccttctgatgt | actccatgcaattagctgcca | aaggcctttgagccctactt |
| SPNS2 | ttactggctccagcgtga | tgatcatgcccaggacag | ctgggcattgcgggtgtc |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeya Paul, J.; Weigel, C.; Müller, T.; Heller, R.; Spiegel, S.; Gräler, M.H. Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate. Cells 2020, 9, 928. https://doi.org/10.3390/cells9040928
Jeya Paul J, Weigel C, Müller T, Heller R, Spiegel S, Gräler MH. Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate. Cells. 2020; 9(4):928. https://doi.org/10.3390/cells9040928
Chicago/Turabian StyleJeya Paul, Jefri, Cynthia Weigel, Tina Müller, Regine Heller, Sarah Spiegel, and Markus H. Gräler. 2020. "Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate" Cells 9, no. 4: 928. https://doi.org/10.3390/cells9040928
APA StyleJeya Paul, J., Weigel, C., Müller, T., Heller, R., Spiegel, S., & Gräler, M. H. (2020). Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate. Cells, 9(4), 928. https://doi.org/10.3390/cells9040928