
Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3β) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and HFD Treatment
2.2. Body Composition
2.3. Echocardiography
2.4. Histology
2.5. Oral Glucose Tolerance Test
2.6. Insulin Tolerance Test
2.7. Sample Preparation and Immunoblotting
2.8. Antibodies
2.9. Statistical Analysis
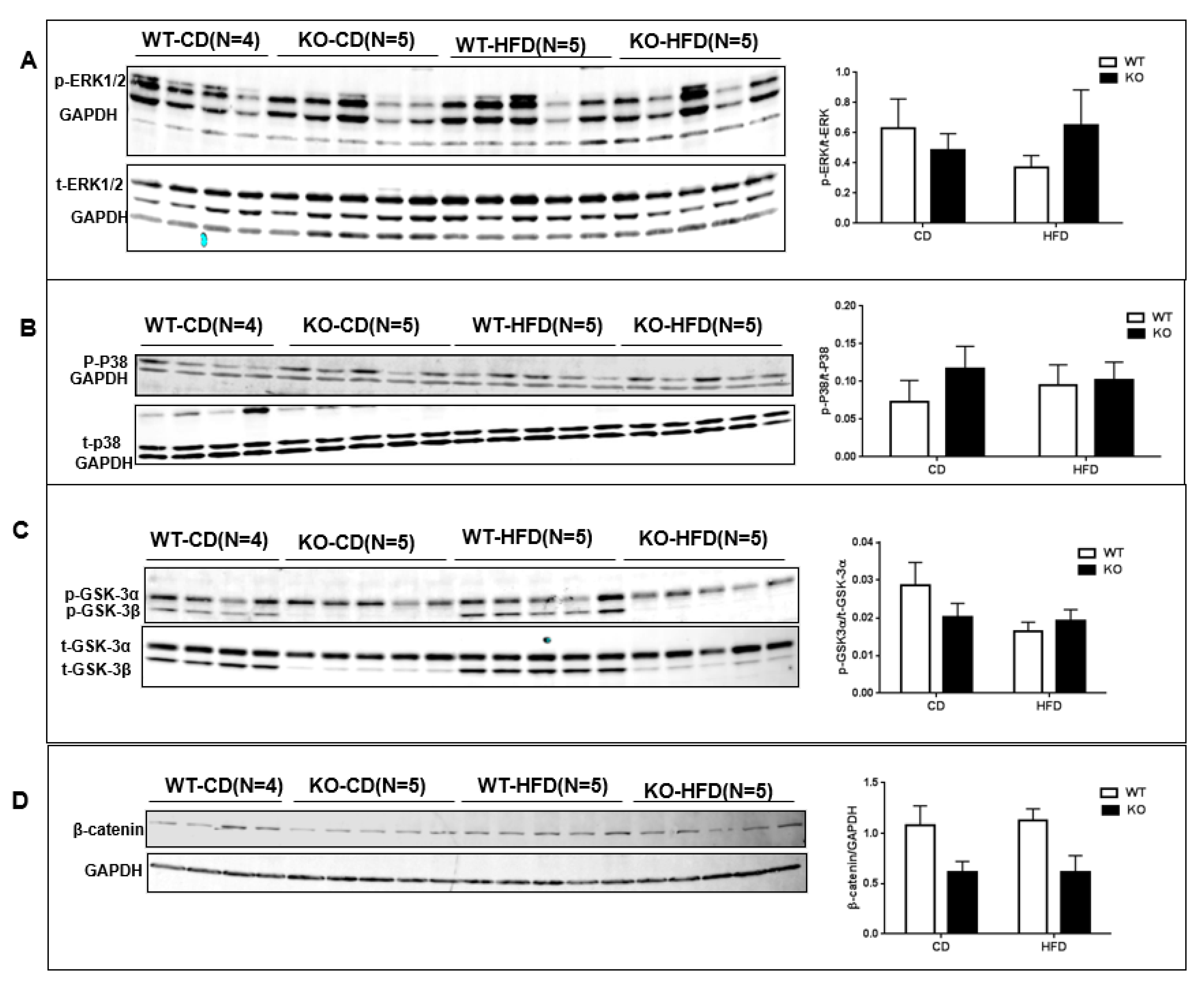
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANP | Atrial natriuretic peptide |
| BNP | Brain natriuretic peptide |
| CD | Control diet |
| CF | Cardiac fibroblast |
| CM | Cardiomyocyte |
| CVD | Cardiovascular diseases |
| EF | Ejection fraction |
| FS | Fractional shortening |
| GSK-3 | Glycogen Synthase Kinase-3 |
| HFD | High-fat diet |
| HW | Heart weight |
| IP | Intraperitoneal injection |
| LVIDd | LV interior dimension -diastolic |
| LVIDs | LV interior dimension -systolic |
| MI | Myocardial infarction |
| TAC | Transverse Aortic Constriction |
| TL | Tibia length |
References
- Lavie, C.J.; Arena, R.; Alpert, M.A.; Milani, R.V.; Ventura, H.O. Management of cardiovascular diseases in patients with obesity. Nat. Rev. Cardiol. 2017, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Willett, W.C.; Hu, F.B. Global obesity: Trends, risk factors and policy implications. Nat. Rev. Endocrinol. 2012, 9, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskin, K.K.; Grueter, C.E.; Kusminski, C.M.; Holland, W.L.; Bookout, A.L.; Satapati, S.; Kong, Y.M.; Burgess, S.C.; Malloy, C.R.; Scherer, P.E.; et al. MED 13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol. Med. 2014, 6, 1610–1621. [Google Scholar] [CrossRef]
- Grueter, C.E.; Van Rooij, E.; Johnson, B.A.; DeLeon, S.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A Cardiac MicroRNA Governs Systemic Energy Homeostasis by Regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Gupte, M.; Tumuluru, S.; Sui, J.Y.; Singh, A.P.; Umbarkar, P.; Parikh, S.; Ahmad, F.; Zhang, Q.; Force, T.; Lal, H. Cardiomyocyte-specific deletion of GSK-3β leads to cardiac dysfunction in a diet induced obesity model. Int. J. Cardiol. 2018, 259, 145–152. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Woodgett, J.R.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Guo, Y.; Gupte, M.; Umbarkar, P.; Singh, A.P.; Sui, J.Y.; Force, T.; Lal, H. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. J. Mol. Cell. Cardiol. 2017, 110, 109–120. [Google Scholar] [CrossRef]
- Chen, X.; Wang, R.; Liu, X.; Wu, Y.; Zhou, T.; Yang, Y.; Perez, A.; Chen, Y.-C.; Hu, L.; Chadarevian, J.P.; et al. A Chemical-Genetic Approach Reveals the Distinct Roles of GSK3α and GSK3β in Regulating Embryonic Stem Cell Fate. Dev. Cell 2017, 43, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Woulfe, K.C.; Gao, E.; Lal, H.; Harris, D.; Fan, Q.; Vagnozzi, R.; DeCaul, M.; Shang, X.; Patel, S.; Woodgett, J.R.; et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ. Res. 2010, 106, 1635–1645. [Google Scholar] [CrossRef]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.R.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Lal, H.; Zhou, J.; Vagnozzi, R.J.; Yu, J.E.; Shang, X.; Woodgett, J.R.; Gao, E.; Force, T. Cardiomyocyte-specific deletion of Gsk3α mitigates post-myocardial infarction remodeling, contractile dysfunction, and heart failure. J. Am. Coll. Cardiol. 2014, 64, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, H.; Zhou, J.; Ahmad, F.; Zaka, R.; Vagnozzi, R.J.; DeCaul, M.; Woodgett, J.R.; Gao, E.; Force, T. Glycogen Synthase Kinase-3α Limits Ischemic Injury, Cardiac Rupture, Post–Myocardial Infarction Remodeling and Death. Circulation 2011, 125, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Doble, B.W.; Macaulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-Specific Role of Glycogen Synthase Kinase 3β in Glucose Homeostasis and Insulin Action. Mol. Cell. Boil. 2008, 28, 6314–6328. [Google Scholar] [CrossRef] [Green Version]
- Macaulay, K.; Woodgett, J.R. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert Opin. Ther. Targets 2008, 12, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tanabe, K.; Baronnier, D.; Patel, S.; Woodgett, J.R.; Cras-Méneur, C.; Permutt, M.A. Conditional ablation of Gsk-3β in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetology 2010, 53, 2600–2610. [Google Scholar] [CrossRef] [Green Version]
- Benzler, J.; Ganjam, G.K.; Kruger, M.; Pinkenburg, O.; Kutschke, M.; Stöhr, S.; Steger, J.; Koch, C.E.; Olkrug, R.; Schwartz, M.W.; et al. Hypothalamic glycogen synthase kinase 3β has a central role in the regulation of food intake and glucose metabolism. Biochem. J. 2012, 447, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.K. Glycogen synthase kinase 3β helps heart to pump better in obese patients. Int. J. Cardiol. 2018, 259, 166–167. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.K.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef]
- Wang, Z.; Li, L.; Zhao, H.; Peng, S.; Zuo, Z. Chronic high fat diet induces cardiac hypertrophy and fibrosis in mice. Metabolism 2015, 64, 917–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Sadoshima, J. Heart over mind: metabolic control of white adipose tissue and liver. EMBO Mol. Med. 2014, 6, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Woodall, B.P.; Gresham, K.S.; Woodall, M.A.; Valenti, M.-C.; Cannavo, A.; Pfleger, J.; Chuprun, J.K.; Drosatos, K.; Koch, W.J. Alteration of myocardial GRK2 produces a global metabolic phenotype. JCI Insight 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Lee, K.-S.; Lee, G.Y.; Jin, H.; Durrance, E.S.; Park, H.S.; Choi, S.H.; Park, K.S.; Kim, Y.-B.; Jang, H.C.; et al. Anti-diabetic efficacy of KICG1338, a novel glycogen synthase kinase-3β inhibitor, and its molecular characterization in animal models of type 2 diabetes and insulin resistance. Mol. Cell. Endocrinol. 2015, 409, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Long-Term Treatment with Novel Glycogen Synthase Kinase-3 Inhibitor Improves Glucose Homeostasis in ob/ob Mice: Molecular Characterization in Liver and Muscle. J. Pharmacol. Exp. Ther. 2005, 316, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Carter, L.; Johnson, K.; Henry, R.R. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes 2002, 51, 2190–2198. [Google Scholar] [CrossRef] [Green Version]
- Eldar-Finkelman, H.; Kaidanovich, O.; Kaidanovich-Beilin, O. The role of glycogen synthase kinase-3 in insulin resistance and Type 2 diabetes. Expert Opin. Ther. Targets 2002, 6, 555–561. [Google Scholar] [CrossRef]
- Matsuda, T.; Zhai, P.; Maejima, Y.; Hong, C.; Gao, S.; Tian, B.; Goto, K.; Takagi, H.; Tamamori-Adachi, M.; Kitajima, S.; et al. Distinct roles of GSK-3α and GSK-3β phosphorylation in the heart under pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 20900–20905. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Rameshwar, P.; Sadoshima, J. Distinct Roles of Glycogen Synthase Kinase (GSK)-3α and GSK-3β in Mediating Cardiomyocyte Differentiation in Murine Bone Marrow-derived Mesenchymal Stem Cells*. J. Boil. Chem. 2009, 284, 36647–36658. [Google Scholar] [CrossRef] [Green Version]
- Baurand, A.; Zelarayan, L.C.; Betney, R.; Gehrke, C.; Dunger, S.; Noack, C.; Busjahn, A.; Huelsken, J.; Taketo, M.M.; Birchmeier, W.; et al. -Catenin Downregulation Is Required for Adaptive Cardiac Remodeling. Circ. Res. 2007, 100, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupte, M.; Umbarkar, P.; Singh, A.P.; Zhang, Q.; Tousif, S.; Lal, H. Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3β) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity. Cells 2020, 9, 1120. https://doi.org/10.3390/cells9051120
Gupte M, Umbarkar P, Singh AP, Zhang Q, Tousif S, Lal H. Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3β) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity. Cells. 2020; 9(5):1120. https://doi.org/10.3390/cells9051120
Chicago/Turabian StyleGupte, Manisha, Prachi Umbarkar, Anand Prakash Singh, Qinkun Zhang, Sultan Tousif, and Hind Lal. 2020. "Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3β) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity" Cells 9, no. 5: 1120. https://doi.org/10.3390/cells9051120
APA StyleGupte, M., Umbarkar, P., Singh, A. P., Zhang, Q., Tousif, S., & Lal, H. (2020). Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3β) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity. Cells, 9(5), 1120. https://doi.org/10.3390/cells9051120