hsa-miR-20b-5p and hsa-miR-363-3p Affect Expression of PTEN and BIM Tumor Suppressor Genes and Modulate Survival of T-ALL Cells In Vitro

 , , , , ,
, , , , , 
Abstract
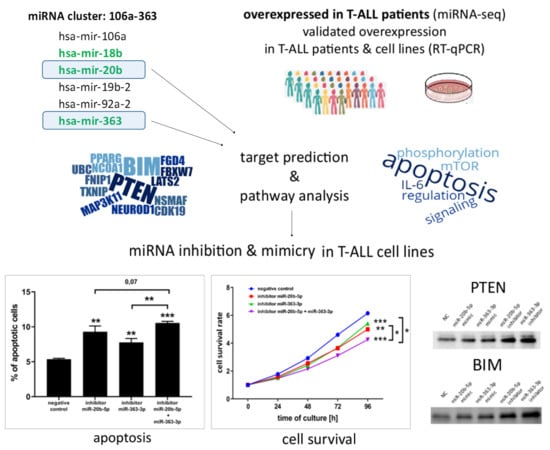
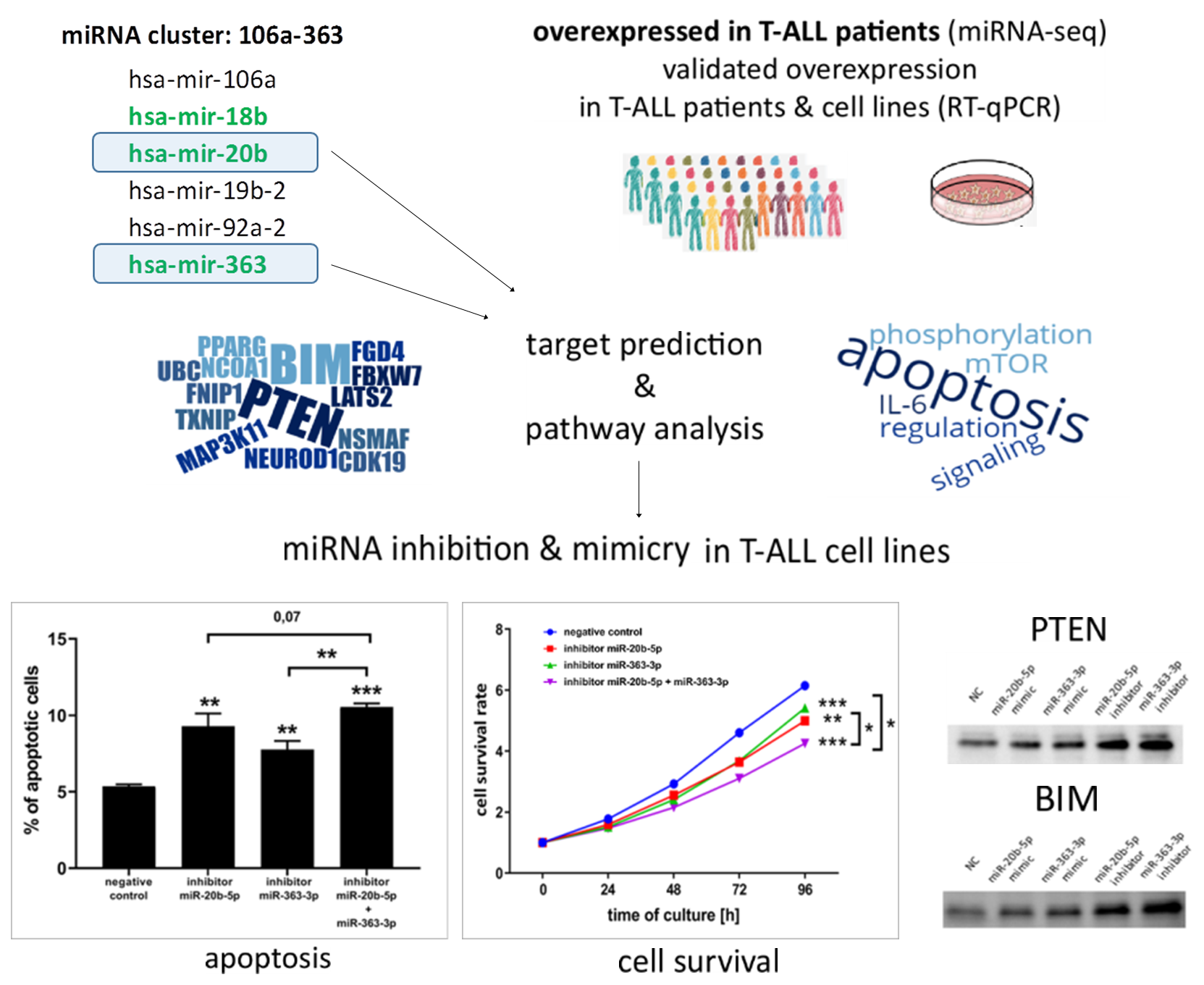
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. miRNA Selection and Target Prediction
2.2. Primary T-ALL Samples and Control Samples
2.3. Cell Lines
2.4. RNA Extraction and RT-qPCR
2.5. Dual Luciferase Reporter Assay
2.6. Transfection of T-ALL Cell Lines
2.7. Western Blotting
2.8. AGO2-RNA Immunoprecipitation and RT-qPCR
2.9. Cell Viability Assay
2.10. Apoptosis Assay
2.11. Cell Cycle Assay
2.12. Statistical Analysis
3. Results
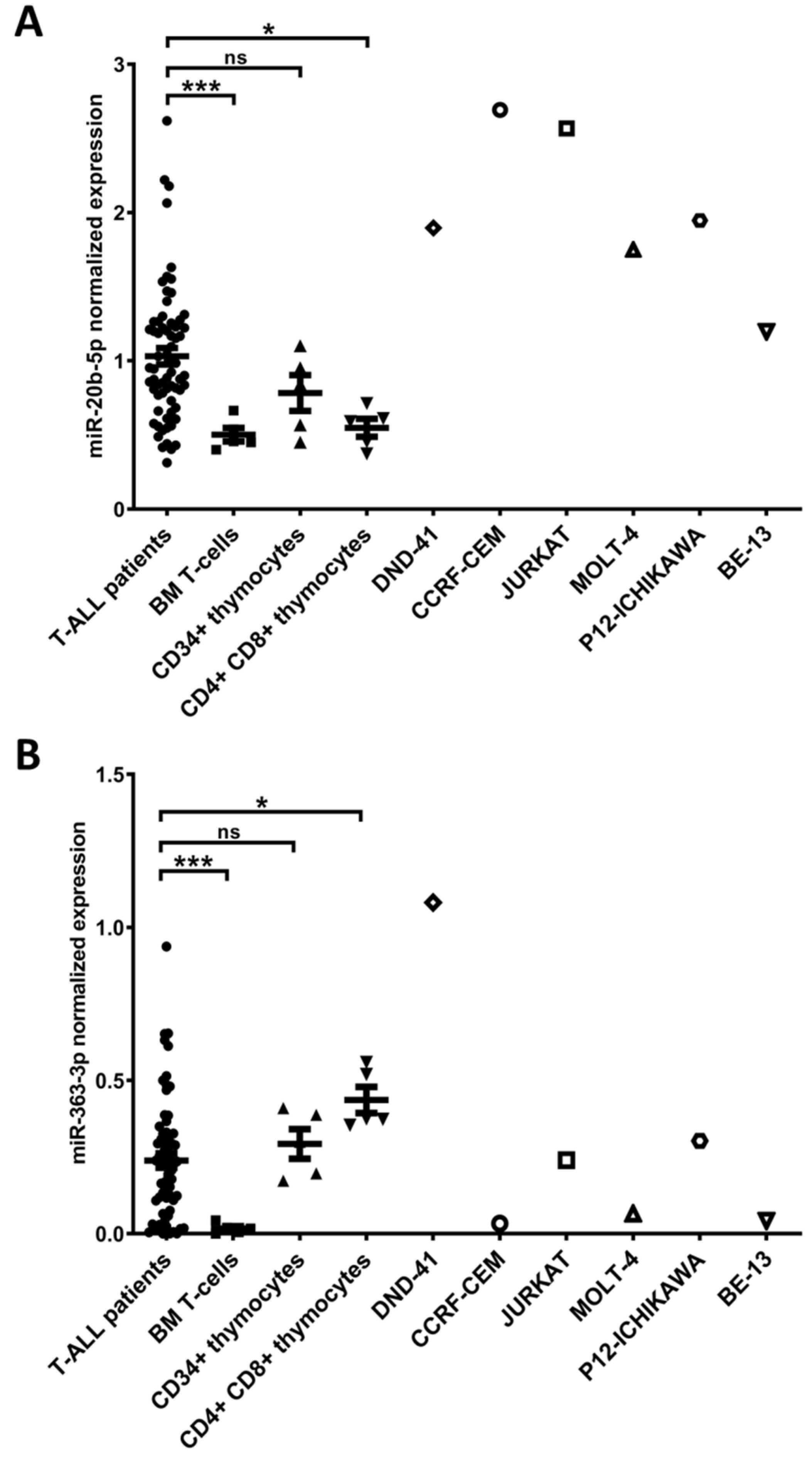
3.1. hsa-miR-20b-5p and hsa-miR-363-3p are Overexpressed in T-ALL and Target Genes Involved in Regulation of Apoptosis
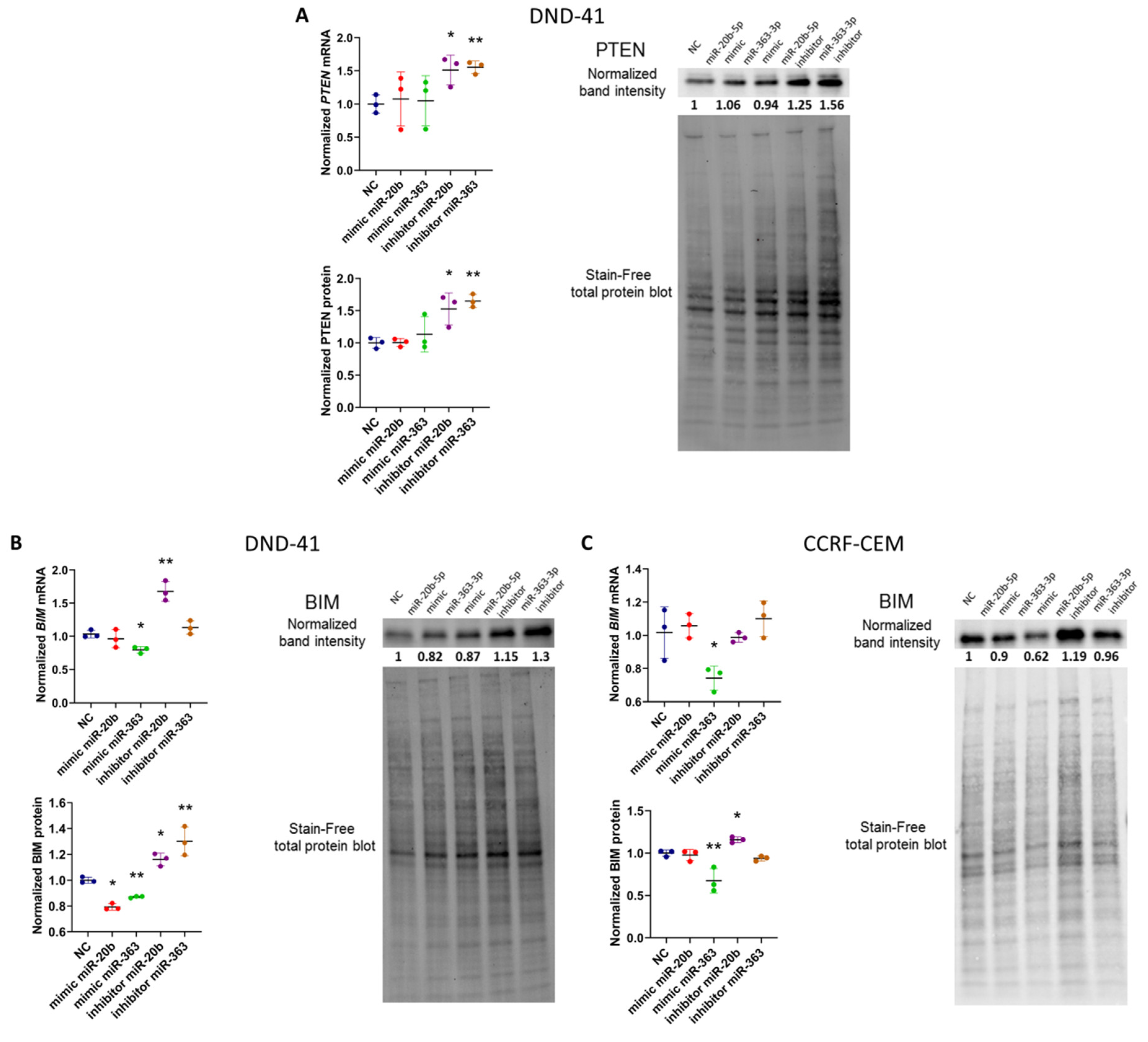
3.2. hsa-miR-20b-5p and hsa-miR-363-3p Downregulate PTEN and BIM in T-ALL Cells In Vitro
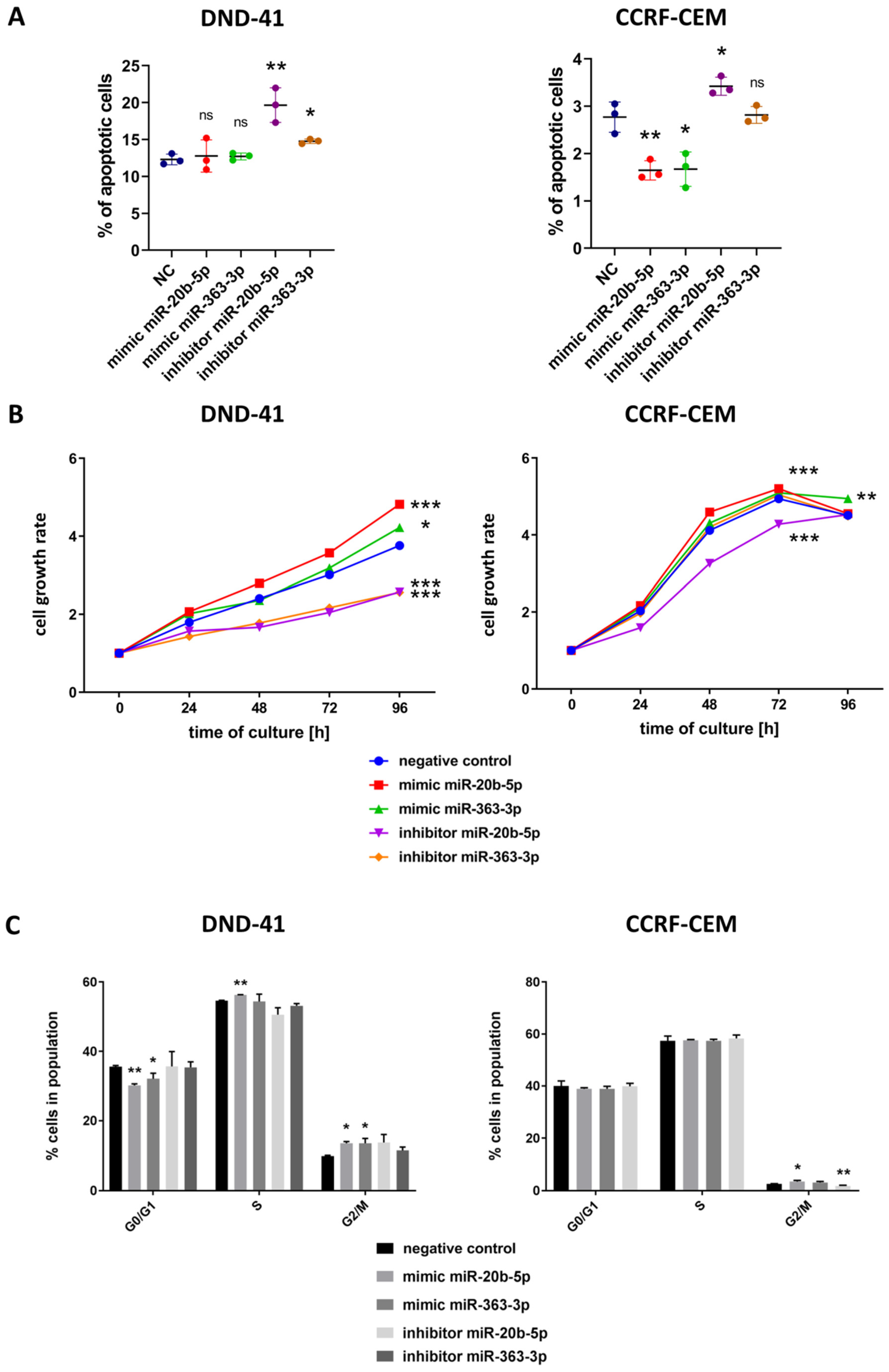
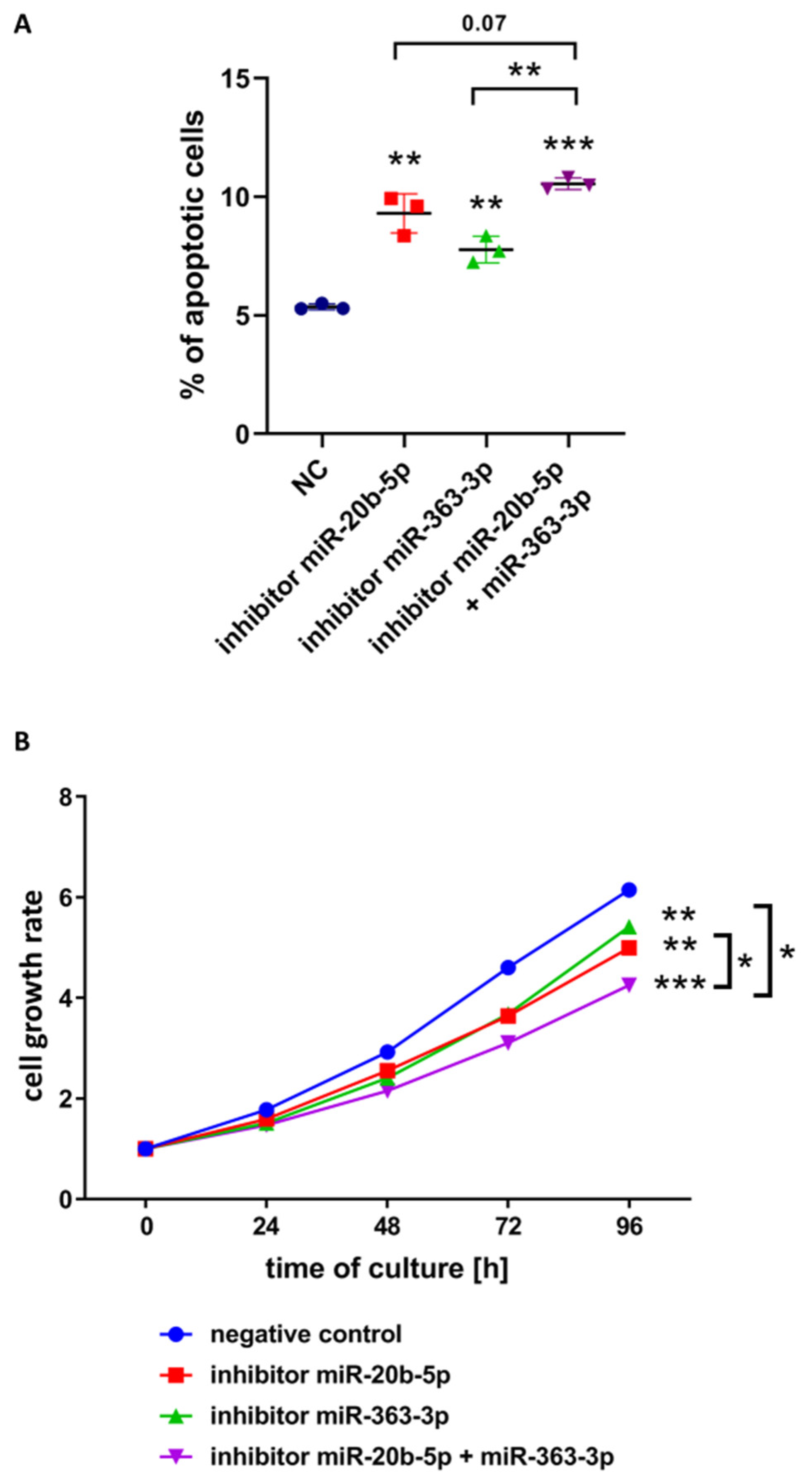
3.3. hsa-miR-20b-5p and hsa-miR-363-3p Exert Anti-Apoptotic and Pro-Proliferative Effects in T-ALL Cells In Vitro
4. Discussion
Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Smedt, R.; Morscio, J.; Goossens, S.; Van Vlierberghe, P. Targeting steroid resistance in T-cell acute lymphoblastic leukemia. Blood Rev. 2019, 39, 100591. [Google Scholar] [CrossRef] [PubMed]
- Drobna, M.; Szarzyńska-Zawadzka, B.; Dawidowska, M. T-cell acute lymphoblastic leukemia from miRNA perspective: Basic concepts, experimental approaches, and potential biomarkers. Blood Rev. 2018, 32, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Kraszewska, M.D.; Dawidowska, M.; Larmonie, N.S.D.; Kosmalska, M.; Sedek, L.; Szczepaniak, M.; Grzeszczak, W.; Langerak, A.W.; Szczepanski, T.; Witt, M.; et al. DNA methylation pattern is altered in childhood T-cell acute lymphoblastic leukemia patients as compared with normal thymic subsets: Insights into CpG island methylator phenotype in T-ALL. Leukemia 2012, 26, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Kraszewska, M.D.; Dawidowska, M.; Szczepański, T.; Witt, M. T-cell acute lymphoblastic leukaemia: Recent molecular biology findings. Br. J. Haematol. 2012, 156, 303–315. [Google Scholar] [CrossRef]
- Durinck, K.; Goossens, S.; Peirs, S.; Wallaert, A.; Van Loocke, W.; Matthijssens, F.; Pieters, T.; Milani, G.; Lammens, T.; Rondou, P.; et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp. Hematol. 2015, 43, 625–639. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, L.; Zhong, M.-L.; Li, J.-F.; Li, B.-S.; Peng, L.-J.; Dai, Y.-T.; Cui, B.-W.; Yan, T.-Q.; Zhang, W.-N.; et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2018, 115, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Gianfelici, V.; Chiaretti, S.; Demeyer, S.; Di Giacomo, F.; Messina, M.; La Starza, R.; Peragine, N.; Paoloni, F.; Geerdens, E.; Pierini, V.; et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica 2016, 101, 941–950. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Wallaert, A.; Van Loocke, W.; Hernandez, L.; Taghon, T.; Speleman, F.; Van Vlierberghe, P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci. Rep. 2017, 7, 7901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawidowska, M.; Jaksik, R.; Drobna, M.; Szarzyńska-Zawadzka, B.; Kosmalska, M.; Sędek, Ł.; Machowska, L.; Lalik, A.; Lejman, M.; Ussowicz, M.; et al. Comprehensive Investigation of miRNome Identifies Novel Candidate miRNA-mRNA Interactions Implicated in T-Cell Acute Lymphoblastic Leukemia. Neoplasia 2019, 21, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Dzikiewicz-Krawczyk, A.; Kok, K.; Slezak-Prochazka, I.; Robertus, J.L.; Bruining, J. ZDHHC11 and ZDHHC11B are critical novel components of the oncogenic MYC-miR-150-MYB network in Burkitt lymphoma. Leukemia 2017, 31, 1470–1473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, S.; Wang, H.; Cao, J.; Huang, X.; Chen, Z.; Xu, P.; Sun, G.; Xu, J.; Lv, J.; et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol. Cancer 2019, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrakis, K.J.; Wolfe, A.L.; Oricchio, E.; Palomero, T.; de Keersmaecker, K.; McJunkin, K.; Zuber, J.; James, T.; Chang, K.; Khan, A.A.; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2010, 12, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Drobna, M.; Szarzyńska-Zawadzka, B.; Daca-Roszak, P.; Kosmalska, M.; Jaksik, R.; Witt, M.; Dawidowska, M. Identification of Endogenous Control miRNAs for RT-qPCR in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Taghon, T.; Waegemans, E.; Van de Walle, I. Notch signaling during human T cell development. Curr. Top. Microbiol. Immunol. 2012, 360, 75–97. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Squiban, B.; Ahmed, S.T.; Frazer, J.K. Creation of a human T-ALL cell line online database. Leuk. Lymphoma 2017, 58, 2728–2730. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Saxena, S.; Singh, B.K.; Kakkar, P. BH3-only protein BIM: An emerging target in chemotherapy. Eur. J. Cell Biol. 2017, 96, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Dominguez, M.; Ferrando, A.A. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle Georget. Tex 2008, 7, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.H.; Bhadri, V.A.; Beck, D.; Thoms, J.A.I.; Yakob, N.A.; Wong, J.W.H.; Knezevic, K.; Pimanda, J.E.; Lock, R.B. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood 2015, 125, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.G.; Kang, S.; Zhao, L.; Vogt, P.K. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 2005, 5, 921–929. [Google Scholar] [CrossRef]
- Samuels, Y.; Ericson, K. Oncogenic PI3K and its role in cancer. Curr. Opin. Oncol. 2006, 18, 77–82. [Google Scholar] [CrossRef]
- Szarzyńska-Zawadzka, B.; Kunz, J.B.; Sędek, Ł.; Kosmalska, M.; Zdon, K.; Biecek, P.; Bandapalli, O.R.; Kraszewska-Hamilton, M.; Jaksik, R.; Drobna, M.; et al. PTEN abnormalities predict poor outcome in children with T-cell acute lymphoblastic leukemia treated according to ALL IC-BFM protocols. Am. J. Hematol. 2019, 94, E93–E96. [Google Scholar] [CrossRef] [Green Version]
- Tesio, M.; Trinquand, A.; Ballerini, P.; Hypolite, G.; Lhermitte, L.; Petit, A.; Ifrah, N.; Baruchel, A.; Dombret, H.; Macintyre, E.; et al. Age-related clinical and biological features of PTEN abnormalities in T-cell acute lymphoblastic leukaemia. Leukemia 2017, 31, 2594–2600. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Zimmermann, M.; Kox, C.; Stanulla, M.; Schrappe, M.; Ludwig, W.-D.; Koehler, R.; Muckenthaler, M.U.; Kulozik, A.E. NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica 2013, 98, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Paganin, M.; Grillo, M.F.; Silvestri, D.; Scapinello, G.; Buldini, B.; Cazzaniga, G.; Biondi, A.; Valsecchi, M.G.; Conter, V.; Te Kronnie, G.; et al. The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br. J. Haematol. 2018, 182, 705–711. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.; Roderick, J.E.; LaBelle, J.L.; Bird, G.; Mathieu, R.; Bodaar, K.; Colon, D.; Pyati, U.; Stevenson, K.E.; Qi, J.; et al. Repression of BIM mediates survival signaling by MYC and AKT in high-risk T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1819–1827. [Google Scholar] [CrossRef]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/mTOR Inhibitor BEZ235. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 621–632. [Google Scholar]
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Ebinu, J.O.; Stang, S.L.; Teixeira, C.; Bottorff, D.A.; Hooton, J.; Blumberg, P.M.; Barry, M.; Bleakley, R.C.; Ostergaard, H.L.; Stone, J.C. RasGRP links T-cell receptor signaling to Ras. Blood 2000, 95, 3199–3203. [Google Scholar] [CrossRef]
- Pierre, S.; Bats, A.-S.; Coumoul, X. Understanding SOS (Son of Sevenless). Biochem. Pharmacol. 2011, 82, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Kortum, R.L.; Sommers, C.L.; Alexander, C.P.; Pinski, J.M.; Li, W.; Grinberg, A.; Lee, J.; Love, P.E.; Samelson, L.E. Targeted Sos1 deletion reveals its critical role in early T-cell development. Proc. Natl. Acad. Sci. USA 2011, 108, 12407–12412. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Thomson, D.W.; Bracken, C.P.; Szubert, J.M.; Goodall, G.J. On Measuring miRNAs after Transient Transfection of Mimics or Antisense Inhibitors. PLoS ONE 2013, 8, e55214. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-F.; Sheng, L.-L.; Wang, G.; Tian, M.; Zhu, L.-Y.; Zhang, R.; Zhang, J.; Zhu, J.-S. miR-363 promotes proliferation and chemo-resistance of human gastric cancer via targeting of FBW7 ubiquitin ligase expression. Oncotarget 2016, 7, 35284–35292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peer, G.V.; Mets, E.; Claeys, S.; Punt, I.D.; Lefever, S.; Ongenaert, M.; Rondou, P.; Speleman, F.; Mestdagh, P.; Vandesompele, J. A high-throughput 3’ UTR reporter screening identifies microRNA interactomes of cancer genes. PLoS ONE 2018, 13, e0194017. [Google Scholar]
- Olive, V.; Jiang, I.; He, L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 2010, 42, 1348–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuu, C.; Utheim, T.P.; Sehic, A. The Three Paralogous MicroRNA Clusters in Development and Disease, miR-17-92, miR-106a-363, and miR-106b-25. Scientifica (Cairo) 2016, 2016, 1379643. [Google Scholar] [CrossRef] [Green Version]
- Tanzer, A.; Stadler, P.F. Molecular evolution of a microRNA cluster. J. Mol. Biol. 2004, 339, 327–335. [Google Scholar] [CrossRef]
- Dylla, L.; Jedlicka, P. Growth-Promoting Role of the miR-106a∼363 Cluster in Ewing Sarcoma. PLoS ONE 2013, 8, e63032. [Google Scholar] [CrossRef]
- Gruszka, R.; Zakrzewska, M. The Oncogenic Relevance of miR-17-92 Cluster and Its Paralogous miR-106b-25 and miR-106a-363 Clusters in Brain Tumors. Int. J. Mol. Sci. 2018, 19, 879. [Google Scholar] [CrossRef] [Green Version]
- Battistella, M.; Romero, M.; Castro-Vega, L.-J.; Gapihan, G.; Bouhidel, F.; Bagot, M.; Feugeas, J.-P.; Janin, A. The High Expression of the microRNA 17-92 Cluster and its Paralogs, and the Downregulation of the Target Gene PTEN, Is Associated with Primary Cutaneous B-Cell Lymphoma Progression. J. Investig. Dermatol. 2015, 135, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Khuu, C.; Jevnaker, A.-M.; Bryne, M.; Osmundsen, H. An investigation into anti-proliferative effects of microRNAs encoded by the miR-106a-363 cluster on human carcinoma cells and keratinocytes using microarray profiling of miRNA transcriptomes. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Mavrakis, K.J.; Van der Meulen, J.; Wolfe, A.L.; Liu, X.P.; Mets, E.; Taghon, T.; Khan, A.A.; Setti, M.; Rondou, P.; Vandenberghe, P.; et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat. Genet. 2011, 43, 673. [Google Scholar] [CrossRef]
- Landais, S.; Quantin, R.; Rassart, E. Radiation Leukemia Virus Common Integration at the Kis2 Locus: Simultaneous Overexpression of a Novel Noncoding RNA and of the Proximal Phf6 Gene. J. Virol. 2005, 79, 11443–11456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landais, S.; Landry, S.; Legault, P.; Rassart, E. Oncogenic potential of the miR-106-363 cluster and its implication in human T-cell leukemia. Cancer Res. 2007, 67, 5699–5707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, A.M.; Wang, B.B.; Li, L.; Channa, N.; Bartha, G.; Wabl, M. Retroviral activation of the mir-106a microRNA cistron in T lymphoma. Retrovirology 2007, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuppers, D.A.; Schmitt, T.M.; Hwang, H.C.; Samraj, L.; Clurman, B.E.; Fero, M.L. The miR-106a~363Xpcl1 miRNA cluster induces murine T cell lymphoma despite transcriptional activation of the p27Kip1 cell cycle inhibitor. Oncotarget 2017, 8, 50680–50691. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.Y.; Gonzalez-Martin, A.; Miletic, A.V.; Lai, M.; Knight, S.; Sabouri-Ghomi, M.; Head, S.R.; Macauley, M.S.; Rickert, R.C.; Xiao, C. Transfection of microRNA Mimics Should Be Used with Caution. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, S.; Stebbing, J.; Frampton, A.E.; Zagorac, S.; Krell, J.; de Giorgio, A.; Trabulo, S.M.; Nguyen, V.T.M.; Magnani, L.; Feng, H.; et al. TGF-β induces miR-100 and miR-125b but blocks let-7a through LIN28B controlling PDAC progression. Nat. Commun. 2018, 9, 1845. [Google Scholar] [CrossRef]
- Reid, G.; Johnson, T.G.; van Zandwijk, N. Manipulating microRNAs for the Treatment of Malignant Pleural Mesothelioma: Past, Present and Future. Front. Oncol. 2020, 10, 105. [Google Scholar] [CrossRef]
- Caracciolo, D.; Montesano, M.; Altomare, E.; Scionti, F.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. The potential role of miRNAs in multiple myeloma therapy. Expert Rev. Hematol. 2018, 11, 793–803. [Google Scholar] [CrossRef]
- Takahashi, R.-U.; Prieto-Vila, M.; Kohama, I.; Ochiya, T. Development of miRNA-based therapeutic approaches for cancer patients. Cancer Sci. 2019, 110, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Pepe, F.; Balatti, V. Role of Non-Coding RNAs in the Development of Targeted Therapy and Immunotherapy Approaches for Chronic Lymphocytic Leukemia. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drobna, M.; Szarzyńska, B.; Jaksik, R.; Sędek, Ł.; Kuchmiy, A.; Taghon, T.; Van Vlierberghe, P.; Szczepański, T.; Witt, M.; Dawidowska, M. hsa-miR-20b-5p and hsa-miR-363-3p Affect Expression of PTEN and BIM Tumor Suppressor Genes and Modulate Survival of T-ALL Cells In Vitro. Cells 2020, 9, 1137. https://doi.org/10.3390/cells9051137
Drobna M, Szarzyńska B, Jaksik R, Sędek Ł, Kuchmiy A, Taghon T, Van Vlierberghe P, Szczepański T, Witt M, Dawidowska M. hsa-miR-20b-5p and hsa-miR-363-3p Affect Expression of PTEN and BIM Tumor Suppressor Genes and Modulate Survival of T-ALL Cells In Vitro. Cells. 2020; 9(5):1137. https://doi.org/10.3390/cells9051137
Chicago/Turabian StyleDrobna, Monika, Bronisława Szarzyńska, Roman Jaksik, Łukasz Sędek, Anna Kuchmiy, Tom Taghon, Pieter Van Vlierberghe, Tomasz Szczepański, Michał Witt, and Małgorzata Dawidowska. 2020. "hsa-miR-20b-5p and hsa-miR-363-3p Affect Expression of PTEN and BIM Tumor Suppressor Genes and Modulate Survival of T-ALL Cells In Vitro" Cells 9, no. 5: 1137. https://doi.org/10.3390/cells9051137
APA StyleDrobna, M., Szarzyńska, B., Jaksik, R., Sędek, Ł., Kuchmiy, A., Taghon, T., Van Vlierberghe, P., Szczepański, T., Witt, M., & Dawidowska, M. (2020). hsa-miR-20b-5p and hsa-miR-363-3p Affect Expression of PTEN and BIM Tumor Suppressor Genes and Modulate Survival of T-ALL Cells In Vitro. Cells, 9(5), 1137. https://doi.org/10.3390/cells9051137