Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases


 , , , ,
, , , ,  , ,
, ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Immunohistochemical Methods
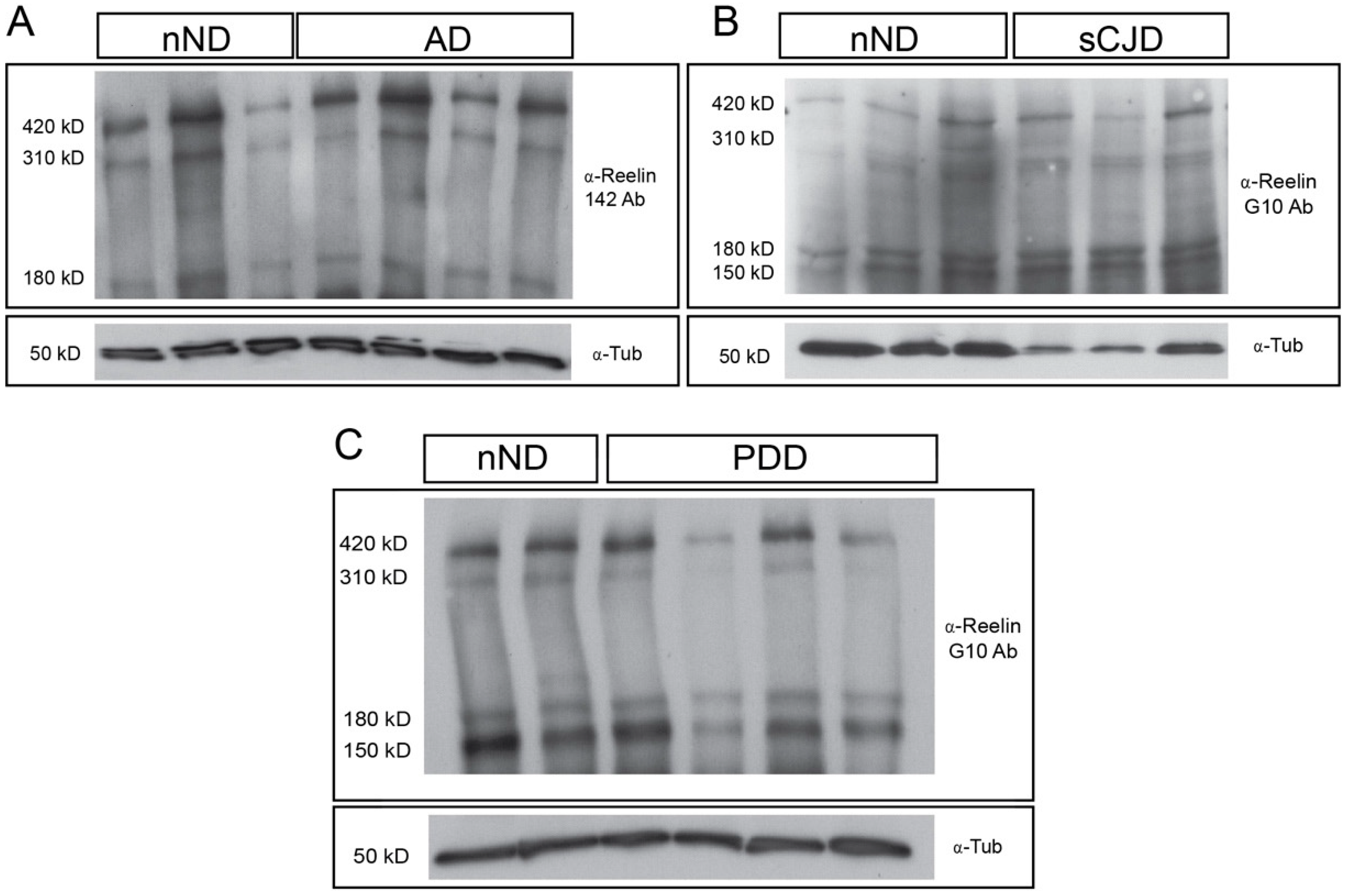
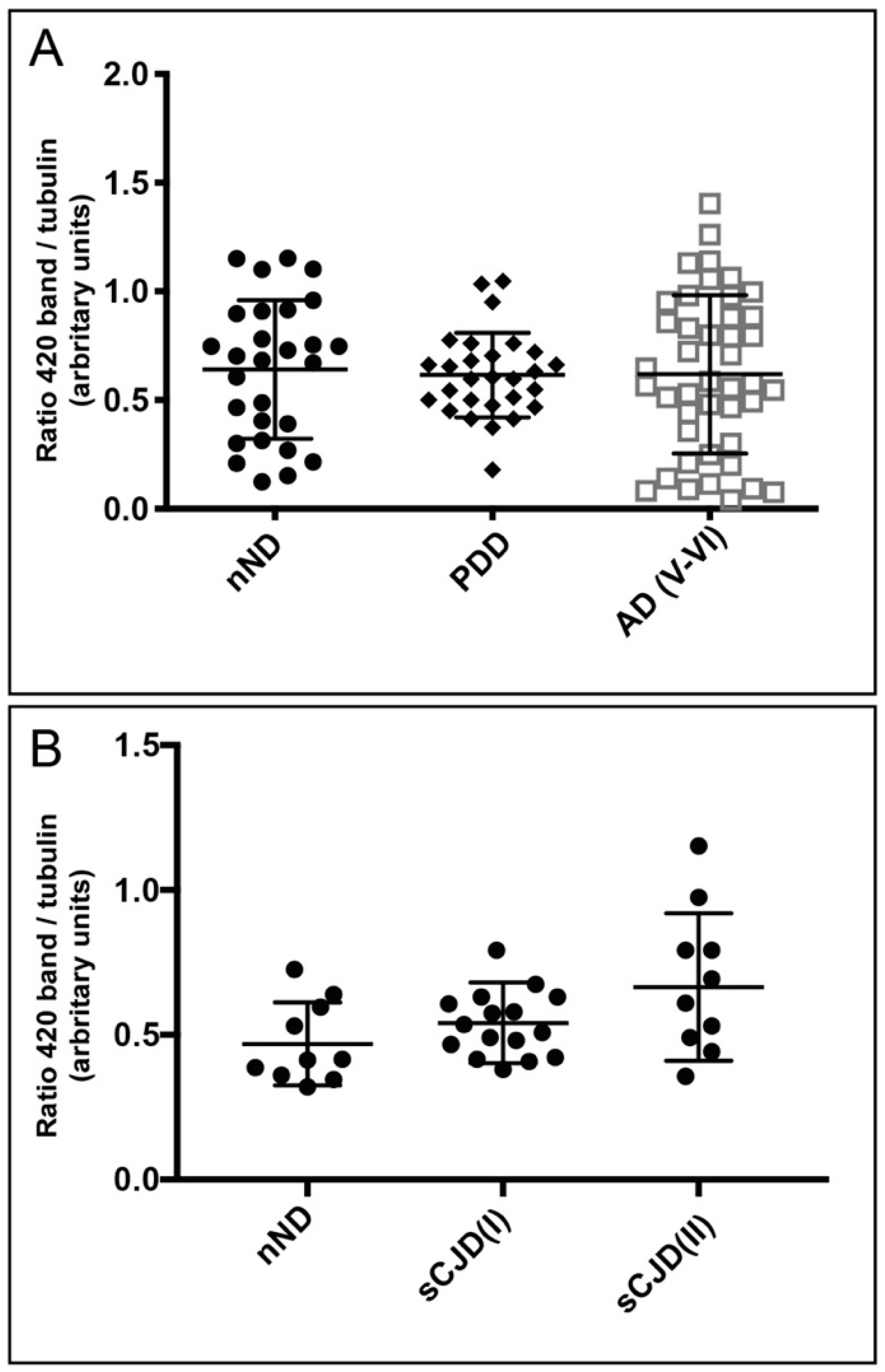
2.3. Western Blotting and Quantification
2.4. Real-Time Quantitative Polimerase Chain Reaction (RT-qPCR)
2.5. Selection Criteria for Reelin Analysis in CSF Samples and RT-qPCR
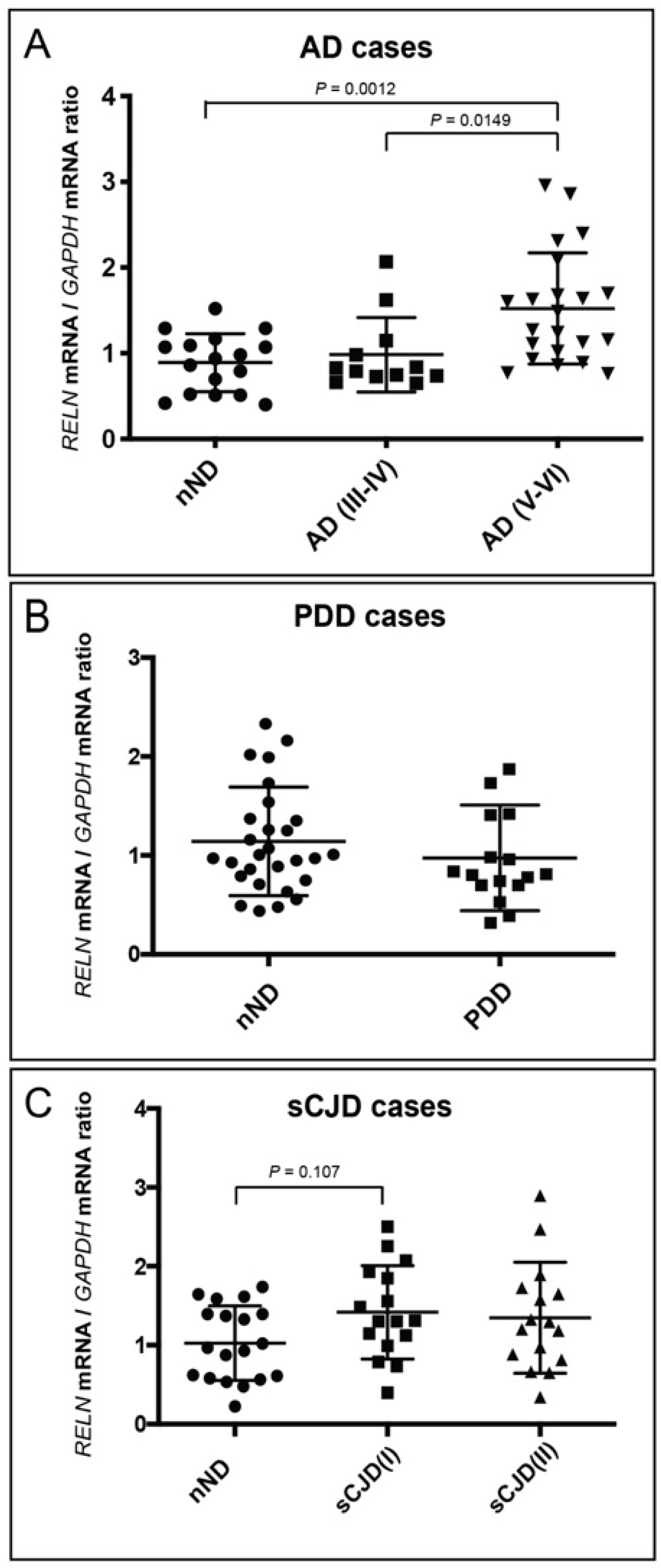
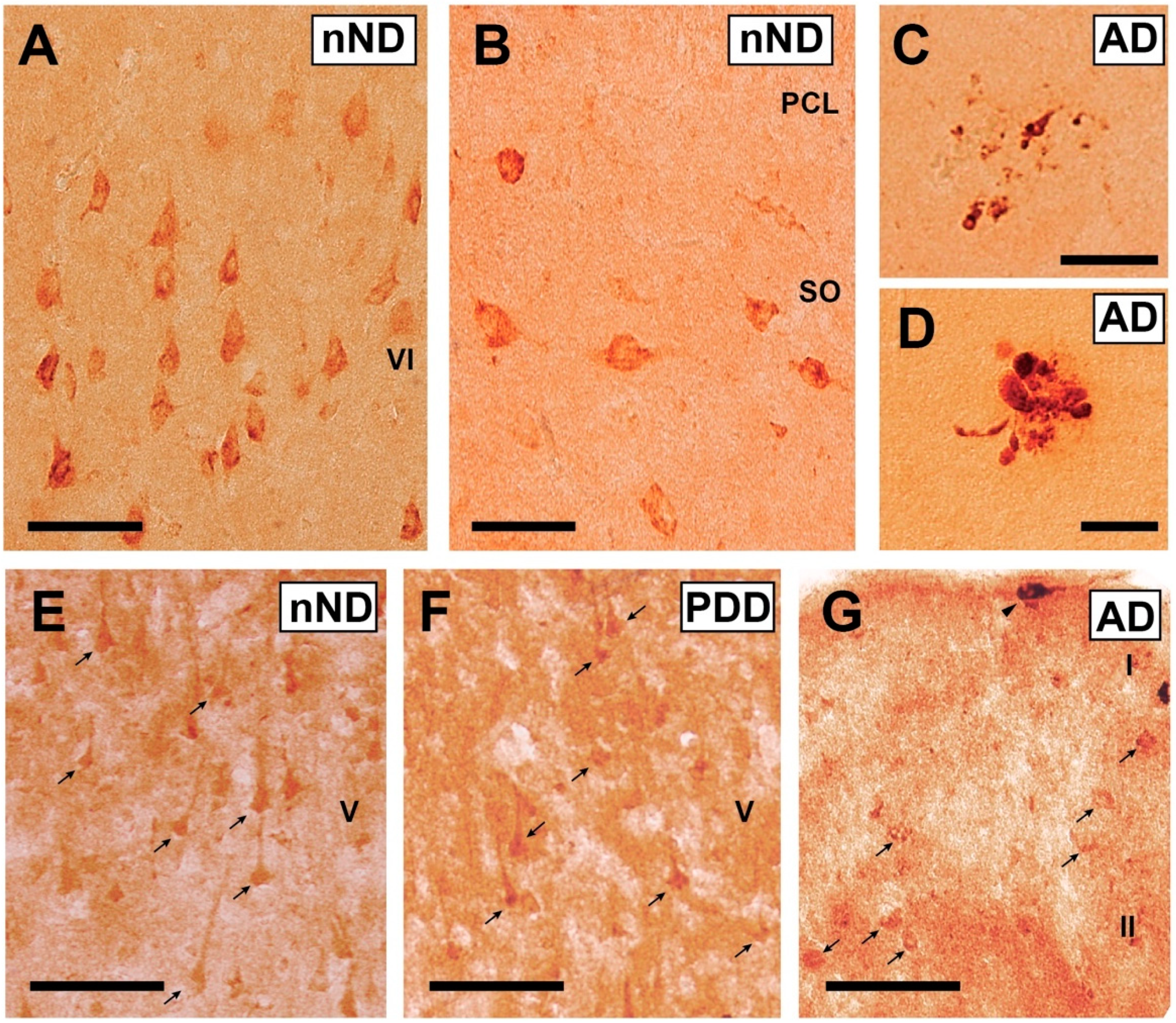
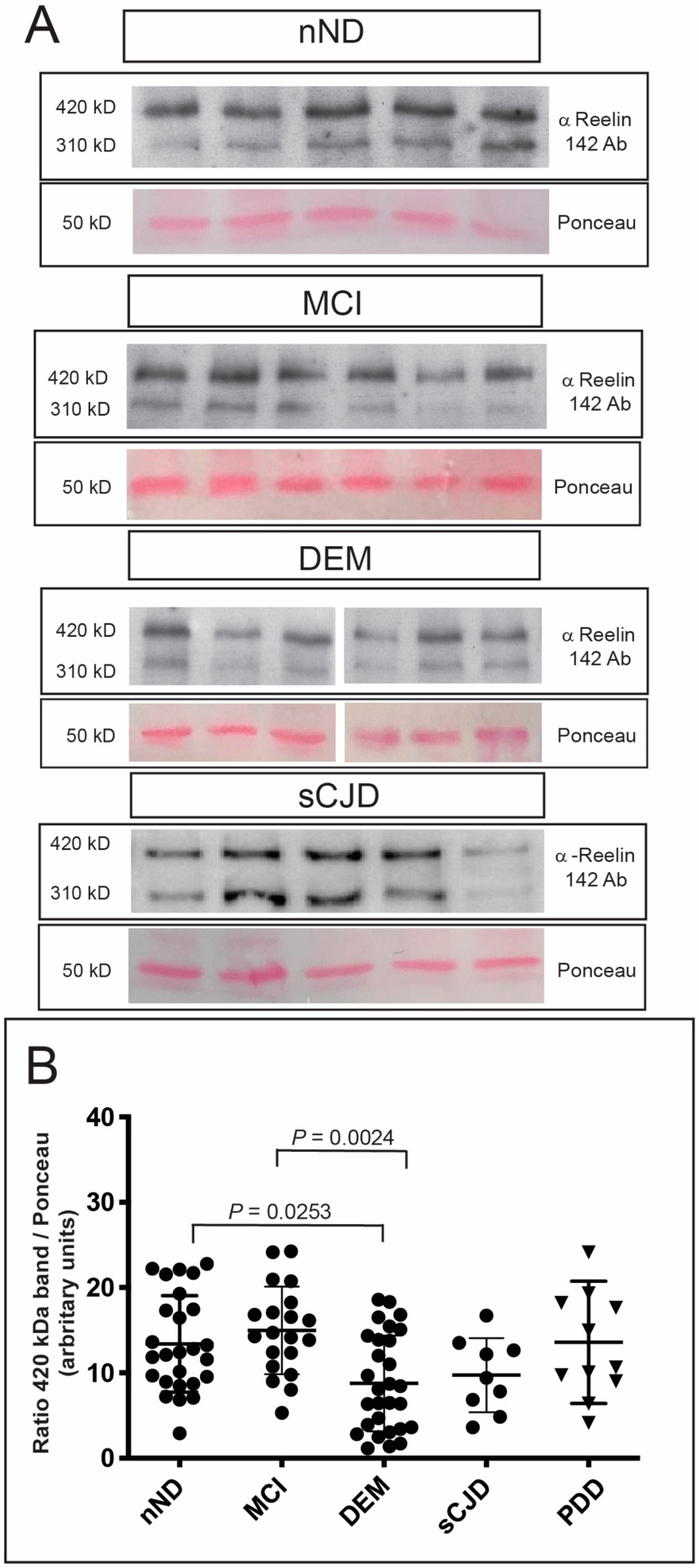
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frotscher, M. Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 2010, 33, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; de Lecea, L.; Martinez, A.; Delgado-Garcia, J.M.; Soriano, E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beffert, U.; Weeber, E.J.; Morfini, G.; Ko, J.; Brady, S.T.; Tsai, L.H.; Sweatt, J.D.; Herz, J. Reelin and cyclin-dependent kinase 5-dependent signals cooperate in regulating neuronal migration and synaptic transmission. J. Neurosci. 2004, 24, 1897–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert de Rouvroit, C.; Bernier, B.; Royaux, I.; de Bergeyck, V.; Goffinet, A.M. Evolutionarily conserved, alternative splicing of reelin during brain development. Exp. Neurol. 1999, 156, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Gui, L.; Goffinet, A.M. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J. Neurosci. 2007, 27, 4243–4252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jossin, Y.; Ignatova, N.; Hiesberger, T.; Herz, J.; Lambert de Rouvroit, C.; Goffinet, A.M. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J. Neurosci. 2004, 24, 514–521. [Google Scholar] [CrossRef]
- Sato, Y.; Kobayashi, D.; Kohno, T.; Kidani, Y.; Prox, J.; Becker-Pauly, C.; Hattori, M. Determination of cleavage site of Reelin between its sixth and seventh repeat and contribution of meprin metalloproteases to the cleavage. J. Biochem. 2016, 159, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Kohno, T.; Nakano, Y.; Suzuki, K.; Ishii, M.; Tagami, H.; Baba, A.; Hattori, M. Mechanism and significance of specific proteolytic cleavage of Reelin. Biochem. Biophys. Res. Commun. 2009, 380, 93–97. [Google Scholar] [CrossRef]
- Tinnes, S.; Ringwald, J.; Haas, C.A. TIMP-1 inhibits the proteolytic processing of Reelin in experimental epilepsy. FASEB J. 2013, 27, 2542–2552. [Google Scholar] [CrossRef]
- Koie, M.; Okumura, K.; Hisanaga, A.; Kamei, T.; Sasaki, K.; Deng, M.; Baba, A.; Kohno, T.; Hattori, M. Cleavage within Reelin repeat 3 regulates the duration and range of the signaling activity of Reelin protein. J. Biol. Chem. 2014, 289, 12922–12930. [Google Scholar] [CrossRef] [Green Version]
- Ogino, H.; Hisanaga, A.; Kohno, T.; Kondo, Y.; Okumura, K.; Kamei, T.; Sato, T.; Asahara, H.; Tsuiji, H.; Fukata, M.; et al. Secreted Metalloproteinase ADAMTS-3 Inactivates Reelin. J. Neurosci. 2017, 37, 3181–3191. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Donath, A.; Steiner, K.M.; Widera, M.P.; Hamzehian, S.; Kanakis, D.; Kolble, K.; ElAli, A.; Hermann, D.M.; Paulus, W.; et al. Reelin depletion is an early phenomenon of Alzheimer’s pathology. J. Alzheimers Dis. 2012, 30, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, N.; Sindic, C.J.; Goffinet, A.M. Characterization of the various forms of the Reelin protein in the cerebrospinal fluid of normal subjects and in neurological diseases. Neurobiol. Dis. 2004, 15, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Notter, T.; Knuesel, I. Reelin immunoreactivity in neuritic varicosities in the human hippocampal formation of non-demented subjects and Alzheimer’s disease patients. Acta Neuropathol. Commun. 2013, 1, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botella-Lopez, A.; Cuchillo-Ibanez, I.; Cotrufo, T.; Mok, S.S.; Li, Q.X.; Barquero, M.S.; Dierssen, M.; Soriano, E.; Saez-Valero, J. Beta-amyloid controls altered Reelin expression and processing in Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, A.; Hatsuta, H.; Kikuchi, M.; Nakaya, A.; Saito, Y.; Tsukie, T.; Hara, N.; Ogishima, S.; Kitamura, N.; Akazawa, K.; et al. Genes associated with the progression of neurofibrillary tangles in Alzheimer’s disease. Transl. Psychiatry 2014, 4, 396. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Font, I.; Iborra-Lazaro, G.; Sanchez-Valle, R.; Molinuevo, J.L.; Cuchillo-Ibanez, I.; Saez-Valero, J. CSF-ApoER2 fragments as a read-out of reelin signaling: Distinct patterns in sporadic and autosomal-dominant Alzheimer disease. Clin. Chim. Acta 2019, 490, 6–11. [Google Scholar] [CrossRef]
- Cuchillo-Ibanez, I.; Balmaceda, V.; Mata-Balaguer, T.; Lopez-Font, I.; Saez-Valero, J. Reelin in Alzheimer’s Disease, Increased Levels but Impaired Signaling: When More is Less. J. Alzheimers Dis. 2016, 52, 403–416. [Google Scholar] [CrossRef]
- Saez-Valero, J.; Costell, M.; Sjogren, M.; Andreasen, N.; Blennow, K.; Luque, J.M. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 132–136. [Google Scholar] [CrossRef]
- Mata, A.; Urrea, L.; Vilches, S.; Llorens, F.; Thune, K.; Espinosa, J.C.; Andreoletti, O.; Sevillano, A.M.; Torres, J.M.; Requena, J.R.; et al. Reelin Expression in Creutzfeldt-Jakob Disease and Experimental Models of Transmissible Spongiform Encephalopathies. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef]
- Alcolea, D.; Clarimon, J.; Carmona-Iragui, M.; Illan-Gala, I.; Morenas-Rodriguez, E.; Barroeta, I.; Ribosa-Nogue, R.; Sala, I.; Sanchez-Saudinos, M.B.; Videla, L.; et al. The Sant Pau Initiative on Neurodegeneration (SPIN) cohort: A data set for biomarker discovery and validation in neurodegenerative disorders. Alzheimers Dement 2019, 5, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Botella-Lopez, A.; Burgaya, F.; Gavin, R.; Garcia-Ayllon, M.S.; Gomez-Tortosa, E.; Pena-Casanova, J.; Urena, J.M.; Del Rio, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.; Ebert, E.; Stoeck, K.; Karch, A.; Collins, S.; Calero, M.; Sklaviadis, T.; Laplanche, J.L.; Golanska, E.; Baldeiras, I.; et al. Validation of 14-3-3 Protein as a Marker in Sporadic Creutzfeldt-Jakob Disease Diagnostic. Mol. Neurobiol. 2016, 53, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Kokmen, E.; Tangelos, E.G. Aging, memory, and mild cognitive impairment. Int. Psychogeriatr. 1997, 9, 65–69. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Zerr, I.; Kallenberg, K.; Summers, D.M.; Romero, C.; Taratuto, A.; Heinemann, U.; Breithaupt, M.; Varges, D.; Meissner, B.; Ladogana, A.; et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009, 132, 2659–2668. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Alvarez, I.; Aguilar, M.; Gonzalez, J.M.; Ysamat, M.; Lorenzo-Bosquet, C.; Alonso, A.; Tartari, J.P.; Romero, S.; Diez-Fairen, M.; Carcel, M.; et al. Clinic-Based Validation of Cerebrospinal Fluid Biomarkers with Florbetapir PET for Diagnosis of Dementia. J. Alzheimers Dis. 2018, 61, 135–143. [Google Scholar] [CrossRef]
- Kobro-Flatmoen, A.; Nagelhus, A.; Witter, M.P. Reelin-immunoreactive neurons in entorhinal cortex layer II selectively express intracellular amyloid in early Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.; Krueger, J.M.; Davis, J.M.; Persico, A.M.; Keller, F.; Smalheiser, N.R. Methodological factors influencing measurement and processing of plasma reelin in humans. BMC Biochem. 2003, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleige, S.; Pfaffl, M.W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Asp. Med. 2006, 27, 126–139. [Google Scholar] [CrossRef]
- Chin, J.; Massaro, C.M.; Palop, J.J.; Thwin, M.T.; Yu, G.Q.; Bien-Ly, N.; Bender, A.; Mucke, L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J. Neurosci. 2007, 27, 2727–2733. [Google Scholar] [CrossRef]
- Krstic, D.; Pfister, S.; Notter, T.; Knuesel, I. Decisive role of Reelin signaling during early stages of Alzheimer’s disease. Neuroscience 2013, 246, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Cerdeno, V.; Galazo, M.J.; Cavada, C.; Clasca, F. Reelin immunoreactivity in the adult primate brain: Intracellular localization in projecting and local circuit neurons of the cerebral cortex, hippocampus and subcortical regions. Cereb. Cortex 2002, 12, 1298–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid beta toxicity in vivo. Sci. Signal. 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujadas, L.; Rossi, D.; Andres, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef]
- Rossi, D.; Gruart, A.; Contreras-Murillo, G.; Muhaisen, A.; Avila, J.; Delgado-Garcia, J.M.; Pujadas, L.; Soriano, E. Reelin reverts biochemical, physiological and cognitive alterations in mouse models of Tauopathy. Prog. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Teixeira, C.M.; Kron, M.M.; Masachs, N.; Zhang, H.; Lagace, D.C.; Martinez, A.; Reillo, I.; Duan, X.; Bosch, C.; Pujadas, L.; et al. Cell-autonomous inactivation of the reelin pathway impairs adult neurogenesis in the hippocampus. J. Neurosci. 2012, 32, 12051–12065. [Google Scholar] [CrossRef] [Green Version]
- Mata-Balaguer, T.; Cuchillo-Ibanez, I.; Calero, M.; Ferrer, I.; Saez-Valero, J. Decreased generation of C-terminal fragments of ApoER2 and increased reelin expression in Alzheimer’s disease. FASEB J. 2018, 32, 3536–3546. [Google Scholar] [CrossRef] [Green Version]
- Cuchillo-Ibanez, I.; Mata-Balaguer, T.; Balmaceda, V.; Arranz, J.J.; Nimpf, J.; Saez-Valero, J. The beta-amyloid peptide compromises Reelin signaling in Alzheimer’s disease. Sci. Rep. 2016, 6, 31646. [Google Scholar] [CrossRef] [Green Version]
- Dayon, L.; Nunez Galindo, A.; Wojcik, J.; Cominetti, O.; Corthesy, J.; Oikonomidi, A.; Henry, H.; Kussmann, M.; Migliavacca, E.; Severin, I.; et al. Alzheimer disease pathology and the cerebrospinal fluid proteome. Alzheimers Res. Ther. 2018, 10, 66. [Google Scholar] [CrossRef]
- Mohammadi, A.; Rashidi, E.; Amooeian, V.G. Brain, blood, cerebrospinal fluid, and serum biomarkers in schizophrenia. Psychiatry Res. 2018, 265, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Ohkubo, N.; Lee, Y.D.; Morishima, A.; Terashima, T.; Kikkawa, S.; Tohyama, M.; Sakanaka, M.; Tanaka, J.; Maeda, N.; Vitek, M.P.; et al. Apolipoprotein E and Reelin ligands modulate tau phosphorylation through an apolipoprotein E receptor/disabled-1/glycogen synthase kinase-3beta cascade. FASEB J. 2003, 17, 295–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lintas, C.; Sacco, R.; Persico, A.M. Differential methylation at the RELN gene promoter in temporal cortex from autistic and typically developing post-puberal subjects. J. Neurodev. Disord. 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Kobow, K.; Jeske, I.; Hildebrandt, M.; Hauke, J.; Hahnen, E.; Buslei, R.; Buchfelder, M.; Weigel, D.; Stefan, H.; Kasper, B.; et al. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 2009, 68, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Grayson, D.R.; Jia, X.; Chen, Y.; Sharma, R.P.; Mitchell, C.P.; Guidotti, A.; Costa, E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 9341–9346. [Google Scholar] [CrossRef] [Green Version]
- Abdolmaleky, H.M.; Cheng, K.H.; Russo, A.; Smith, C.L.; Faraone, S.V.; Wilcox, M.; Shafa, R.; Glatt, S.J.; Nguyen, G.; Ponte, J.F.; et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: A preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 134, 60–66. [Google Scholar] [CrossRef]
- Zetterberg, H. Cerebrospinal fluid total prion protein: A potential in vivo marker of cerebral prion pathology. JAMA Neurol. 2015, 72, 261–263. [Google Scholar] [CrossRef]
- Meyne, F.; Gloeckner, S.F.; Ciesielczyk, B.; Heinemann, U.; Krasnianski, A.; Meissner, B.; Zerr, I. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J. Alzheimers Dis. 2009, 17, 863–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens, F.; Ansoleaga, B.; Garcia-Esparcia, P.; Zafar, S.; Grau-Rivera, O.; Lopez-Gonzalez, I.; Blanco, R.; Carmona, M.; Yague, J.; Nos, C.; et al. PrP mRNA and protein expression in brain and PrP(c) in CSF in Creutzfeldt-Jakob disease MM1 and VV2. Prion 2013, 7, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.; Schlomm, M.; Hasan, B.; Beekes, M.; Mitrova, E.; Korth, C.; Breil, A.; Carimalo, J.; Gawinecka, J.; Varges, D.; et al. Codon 129 polymorphism and the E200K mutation do not affect the cellular prion protein isoform composition in the cerebrospinal fluid from patients with Creutzfeldt-Jakob disease. Eur. J. Neurosci. 2010, 31, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx 2004, 1, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; de Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.A.; Caruncho, H.J.; Costa, E.; Pesold, C.; Liu, W.S.; Guidotti, A. In Patas monkey, glutamic acid decarboxylase-67 and reelin mRNA coexpression varies in a manner dependent on layers and cortical areas. J. Comp. Neurol. 2002, 451, 279–288. [Google Scholar] [CrossRef]
- Martinez-Cerdeno, V.; Galazo, M.J.; Clasca, F. Reelin-immunoreactive neurons, axons, and neuropil in the adult ferret brain: Evidence for axonal secretion of reelin in long axonal pathways. J. Comp. Neurol. 2003, 463, 92–116. [Google Scholar] [CrossRef]
- Impagnatiello, F.; Guidotti, A.R.; Pesold, C.; Dwivedi, Y.; Caruncho, H.; Pisu, M.G.; Uzunov, D.P.; Smalheiser, N.R.; Davis, J.M.; Pandey, G.N.; et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. USA 1998, 95, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.A.; Pesold, C.; Liu, W.S.; Kriho, V.; Guidotti, A.; Pappas, G.D.; Costa, E. Colocalization of integrin receptors and reelin in dendritic spine postsynaptic densities of adult nonhuman primate cortex. Proc. Natl. Acad. Sci. USA 2000, 97, 3550–3555. [Google Scholar] [CrossRef]
- Roberts, R.C.; Xu, L.; Roche, J.K.; Kirkpatrick, B. Ultrastructural localization of reelin in the cortex in post-mortem human brain. J. Comp. Neurol. 2005, 482, 294–308. [Google Scholar] [CrossRef]
- Pesold, C.; Impagnatiello, F.; Pisu, M.G.; Uzunov, D.P.; Costa, E.; Guidotti, A.; Caruncho, H.J. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA 1998, 95, 3221–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudan, A.; Sidler, C.; Knuesel, I. Accumulation of reelin-positive plaques is accompanied by a decline in basal forebrain projection neurons during normal aging. Eur. J. Neurosci. 2009, 30, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Doehner, J.; Madhusudan, A.; Konietzko, U.; Fritschy, J.M.; Knuesel, I. Co-localization of Reelin and proteolytic AbetaPP fragments in hippocampal plaques in aged wild-type mice. J. Alzheimers Dis. 2010, 19, 1339–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knuesel, I.; Nyffeler, M.; Mormede, C.; Muhia, M.; Meyer, U.; Pietropaolo, S.; Yee, B.K.; Pryce, C.R.; LaFerla, F.M.; Marighetto, A.; et al. Age-related accumulation of Reelin in amyloid-like deposits. Neurobiol. Aging. 2009, 30, 697–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Reelin in plaques of beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 316, 145–148. [Google Scholar] [CrossRef]
- Kocherhans, S.; Madhusudan, A.; Doehner, J.; Breu, K.S.; Nitsch, R.M.; Fritschy, J.M.; Knuesel, I. Reduced Reelin expression accelerates amyloid-beta plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J. Neurosci. 2010, 30, 9228–9240. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Tunon, T.; Soriano, E.; del Rio, A.; Iraizoz, I.; Fonseca, M.; Guionnet, N. Calbindin D-28k immunoreactivity in the temporal neocortex in patients with Alzheimer’s disease. Clin. Neuropathol. 1993, 12, 53–58. [Google Scholar]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [Green Version]
- Hornig, T.; Haas, C.; Sturm, L.; Fiebich, B.; Tebartz van Elst, L. Increased Blood-Reelin-Levels in First Episode Schizophrenia. PLoS ONE 2015, 10, 0134671. [Google Scholar] [CrossRef]
- Cuchillo-Ibanez, I.; Andreo-Lillo, P.; Pastor-Ferrandiz, L.; Carratala-Marco, F.; Saez-Valero, J. Elevated Plasma Reelin Levels in Children with Autism. Front. Psychiatry 2020, 11, 242. [Google Scholar] [CrossRef]
- Sturm, L.; van Elst, L.T.; Fiebich, B.; Wolkewitz, M.; Hornig, T. Intra-day variations of blood reelin levels in healthy individuals. Arch. Med. Sci. 2020, 16, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Costa, E.; Guidotti, A.; Impagnatiello, F.; Auta, J.; Lacor, P.; Kriho, V.; Pappas, G.D. Expression of reelin in adult mammalian blood, liver, pituitary pars intermedia, and adrenal chromaffin cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1281–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botella-Lopez, A.; de Madaria, E.; Jover, R.; Bataller, R.; Sancho-Bru, P.; Candela, A.; Compan, A.; Perez-Mateo, M.; Martinez, S.; Saez-Valero, J. Reelin is overexpressed in the liver and plasma of bile duct ligated rats and its levels and glycosylation are altered in plasma of humans with cirrhosis. Int. J. Biochem. Cell Biol. 2008, 40, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Okamura, Y.; Nomoto, S.; Kanda, M.; Hayashi, M.; Nishikawa, Y.; Fujii, T.; Sugimoto, H.; Takeda, S.; Nakao, A. Reduced expression of reelin (RELN) gene is associated with high recurrence rate of hepatocellular carcinoma. Ann. Surg. Oncol. 2011, 18, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Carotti, S.; Perrone, G.; Amato, M.; Vespasiani Gentilucci, U.; Righi, D.; Francesconi, M.; Pellegrini, C.; Zalfa, F.; Zingariello, M.; Picardi, A.; et al. Reelin expression in human liver of patients with chronic hepatitis C infection. Eur. J. Histochem. 2017, 61, 2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ND | RELN in FC | Reelin in FC |
| AD(III-IV) | - | - |
| AD(V-VI) | ↑, ⇑ | - |
| PDD | - | - |
| sCJD(I) | > | > |
| sCJD(II) | - | > |
| DIAG | Reelin in CSF | |
| MCI | - | |
| DEM | ↓, ⇓ | |
| PDD | - | |
| sCJD | < |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lidón, L.; Urrea, L.; Llorens, F.; Gil, V.; Alvarez, I.; Diez-Fairen, M.; Aguilar, M.; Pastor, P.; Zerr, I.; Alcolea, D.; et al. Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases. Cells 2020, 9, 1252. https://doi.org/10.3390/cells9051252
Lidón L, Urrea L, Llorens F, Gil V, Alvarez I, Diez-Fairen M, Aguilar M, Pastor P, Zerr I, Alcolea D, et al. Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases. Cells. 2020; 9(5):1252. https://doi.org/10.3390/cells9051252
Chicago/Turabian StyleLidón, Laia, Laura Urrea, Franc Llorens, Vanessa Gil, Ignacio Alvarez, Monica Diez-Fairen, Miguel Aguilar, Pau Pastor, Inga Zerr, Daniel Alcolea, and et al. 2020. "Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases" Cells 9, no. 5: 1252. https://doi.org/10.3390/cells9051252
APA StyleLidón, L., Urrea, L., Llorens, F., Gil, V., Alvarez, I., Diez-Fairen, M., Aguilar, M., Pastor, P., Zerr, I., Alcolea, D., Lleó, A., Vidal, E., Gavín, R., Ferrer, I., & Del Rio, J. A. (2020). Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases. Cells, 9(5), 1252. https://doi.org/10.3390/cells9051252