Metabolic Adaptation during nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viability Assay and IC50 Calculation
2.3. Establishing Chemotherapy-Resistant Pancreatic Cancer Cell Lines
2.4. Cell Death Assay
2.5. Untargeted GC/MS-Based Metabolic Profiling
2.6. Statistical Analysis
2.7. Data Availability
3. Results and Discussion
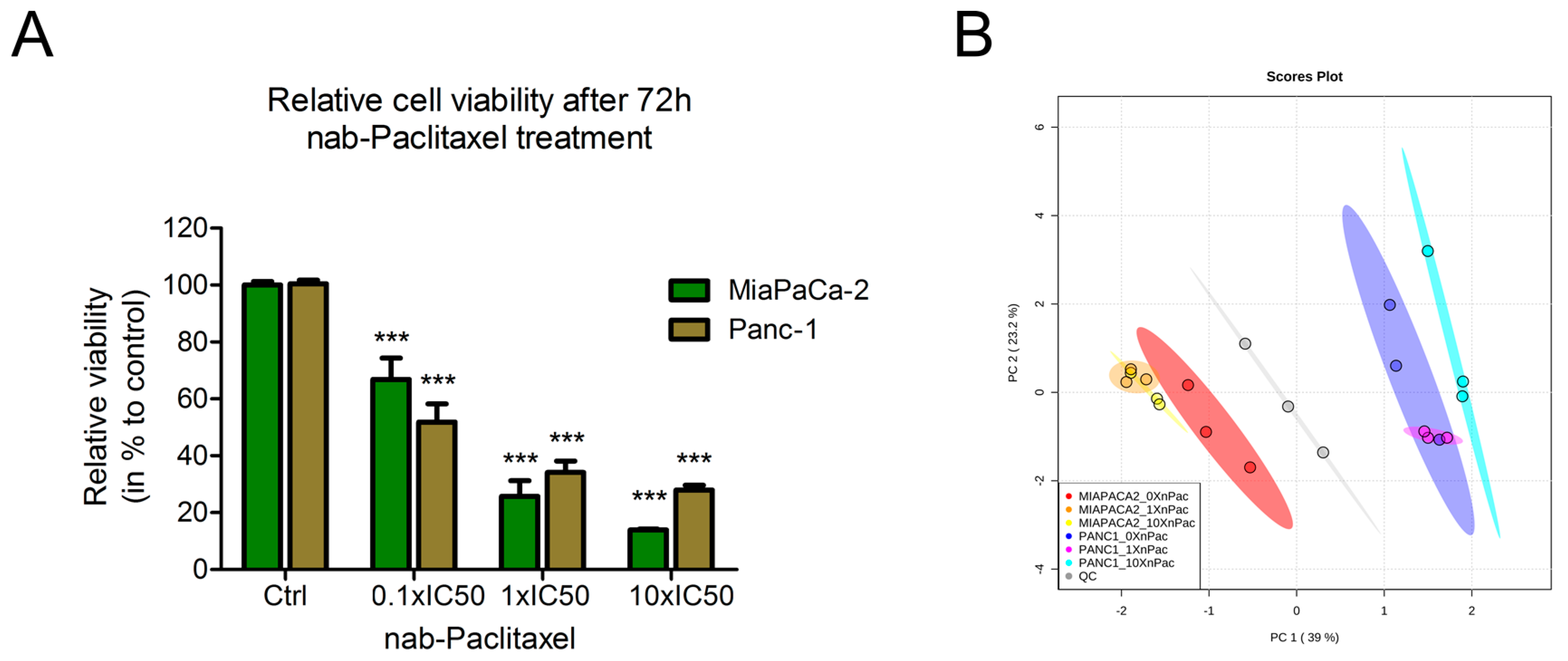
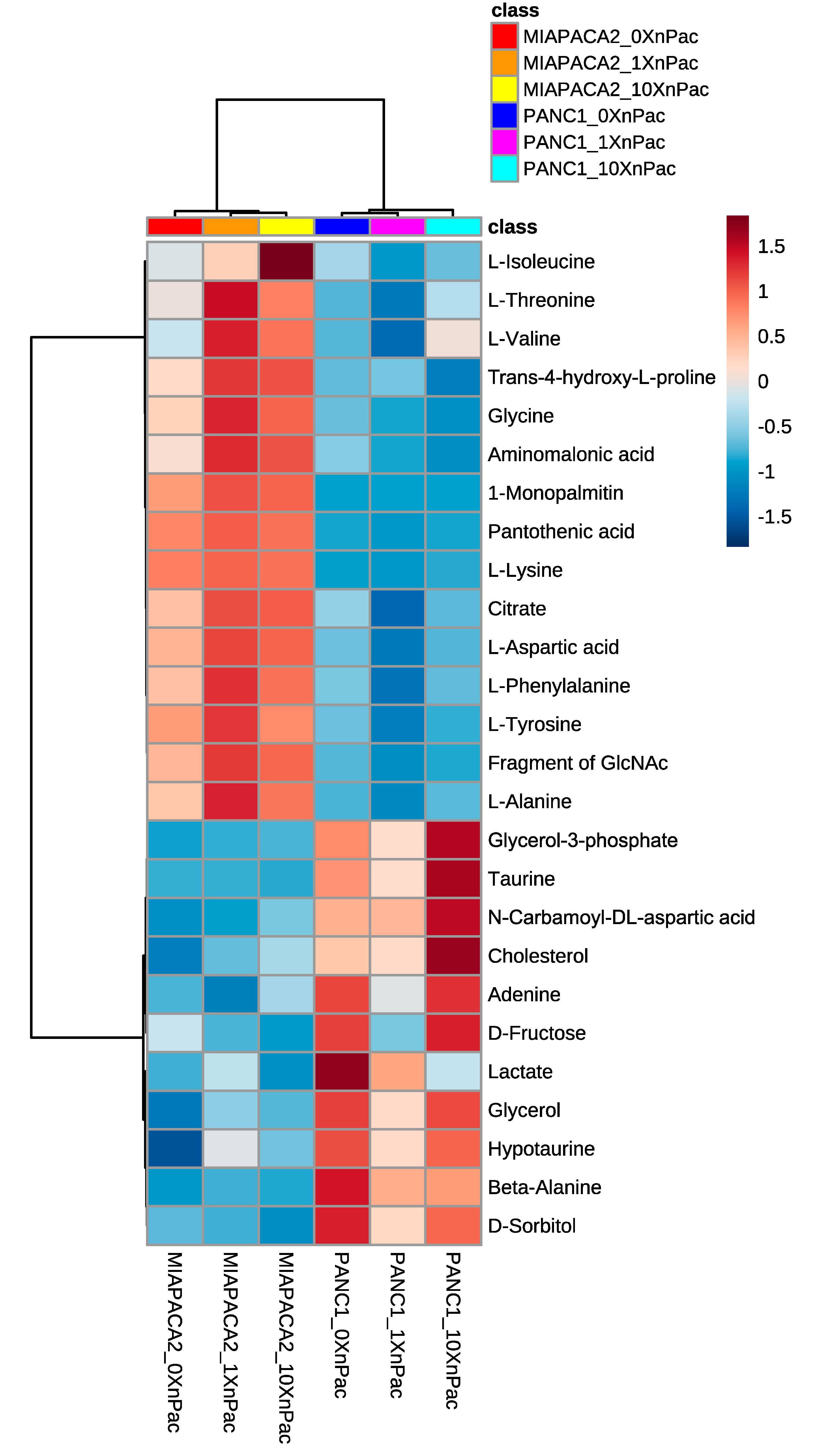
3.1. Treatment of Pancreatic Cancer Cells Lines with nab-Paclitaxel Resulted in Few Metabolic Alterations
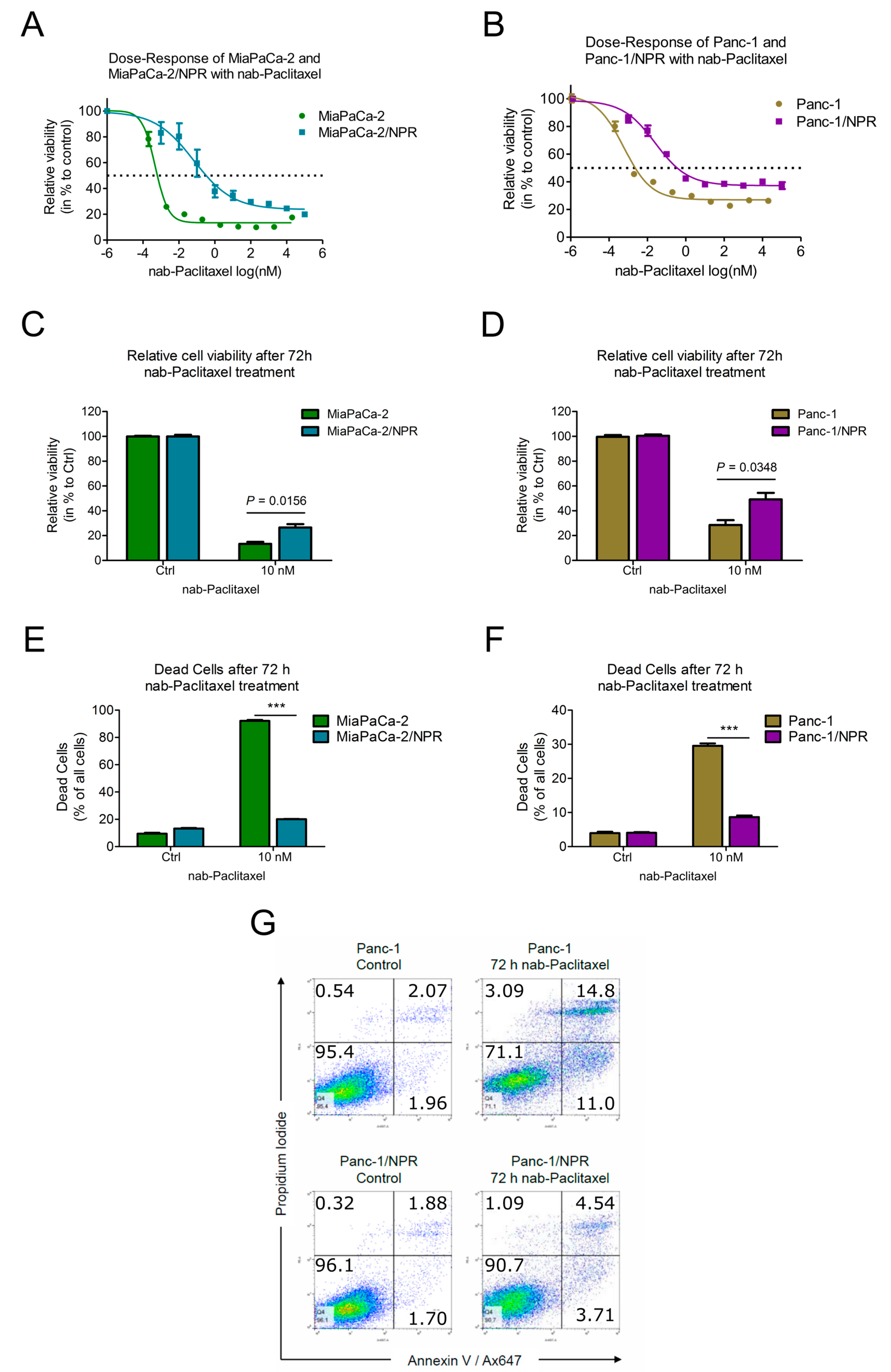
3.2. Chemotherapy-Resistant PDAC Cell Lines Had Higher IC50-Values Compared to Their Parental Cell Lines
3.3. Resistant Cells Showed Less Apoptosis upon Chemotherapy Treatment
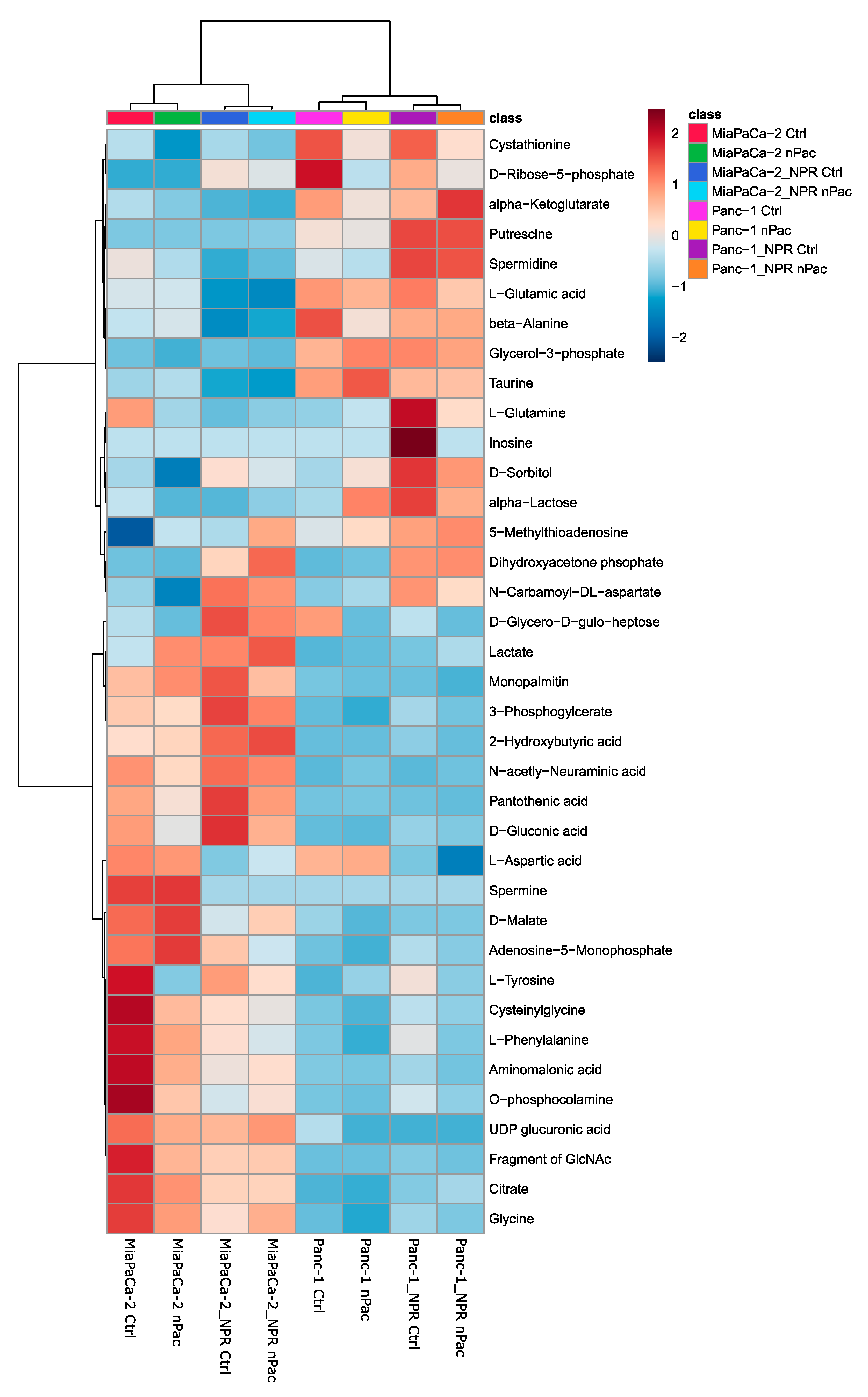
3.4. Metabolic Analysis of Chemotherapy-Resistant PDAC Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muniraj, T.; Jamidar, P.A.; Aslanian, H.R. Pancreatic cancer: A comprehensive review and update. Dis. Mon. DM 2013, 59, 368–402. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- SEER Cancer Statistics Review, 1975–2016. Available online: https://seer.cancer.gov/csr/1975_2016/index.html (accessed on 22 April 2020).
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Ther. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.-M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, S.; Zhang, B.; Ni, Q.; Yu, X.; Xu, J. Circulating biomarkers for early diagnosis of pancreatic cancer: Facts and hopes. Am. J. Cancer Res. 2018, 8, 332–353. [Google Scholar]
- Scarà, S.; Bottoni, P.; Scatena, R. CA 19-9: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 247–260. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Bujanda, L. Non-invasive biomarkers in pancreatic cancer diagnosis: What we need versus what we have. Ann. Transl. Med. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Takai, E.; Yachida, S. Circulating tumor DNA as a liquid biopsy target for detection of pancreatic cancer. World J. Gastroenterol. 2016, 22, 8480–8488. [Google Scholar] [CrossRef]
- Nagayoshi, Y.; Nakamura, M.; Matsuoka, K.; Ohtsuka, T.; Mori, Y.; Kono, H.; Aso, T.; Ideno, N.; Takahata, S.; Ryo, A.; et al. Profiling of autoantibodies in sera of pancreatic cancer patients. Ann. Surg. Oncol. 2014, 21 (Suppl. 3), S459–S465. [Google Scholar] [CrossRef] [PubMed]
- Lagies, S.; Schlimpert, M.; Braun, L.M.; Kather, M.; Plagge, J.; Erbes, T.; Wittel, U.A.; Kammerer, B. Unraveling altered RNA metabolism in pancreatic cancer cells by liquid-chromatography coupling to ion mobility mass spectrometry. Anal. Bioanal. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-B.; Yang, Y.; Zhao, Y.-P.; Zhang, T.-P.; Liao, Q.; Shu, H. Recent studies of 5-fluorouracil resistance in pancreatic cancer. World J. Gastroenterol. WJG 2014, 20, 15682–15690. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Mayer, R.J.; Gonin, R.; Arbuck, S.G. Fluorouracil and high-dose leucovorin in previously untreated patients with advanced adenocarcinoma of the pancreas: Results of a phase II trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1991, 9, 2128–2133. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, V.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Comparison of the cellular pharmacokinetics and toxicity of 2′,2′-difluorodeoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1988, 48, 4024–4031. [Google Scholar]
- Heinemann, V. Gemcitabine: Progress in the Treatment of Pancreatic Cancer. Oncology 2001, 60, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Park, S.W. Systemic chemotherapy in advanced pancreatic cancer. Gut Liver 2016, 10, 340–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, G.; Pancione, M.; Olivieri, N.; Parcesepe, P.; Velocci, M.; Di Raimo, T.; Coppola, L.; Toffoli, G.; D’Andrea, M.R. Nano albumin bound-paclitaxel in pancreatic cancer: Current evidences and future directions. World J. Gastroenterol. 2017, 23, 5875–5886. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Ueno, H.; Kiyosawa, K.; Kaniwa, N. Pharmacogenomics of gemcitabine: Can genetic studies lead to tailor-made therapy? Br. J. Cancer 2007, 97, 145–151. [Google Scholar] [CrossRef]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [Green Version]
- Iovanna, J.; Mallmann, M.C.; Gonçalves, A.; Turrini, O.; Dagorn, J.-C. Current Knowledge on Pancreatic Cancer. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Gnanamony, M.; Gondi, C.S. Chemoresistance in pancreatic cancer: Emerging concepts. Oncol. Lett. 2017, 13, 2507–2513. [Google Scholar] [CrossRef] [Green Version]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updatates 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Maréchal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; Cass, C.E.; Young, J.; Salmon, I.; Devière, J.; et al. Human Equilibrative Nucleoside Transporter 1 and Human Concentrative Nucleoside Transporter 3 Predict Survival after Adjuvant Gemcitabine Therapy in Resected Pancreatic Adenocarcinoma. Clin. Cancer Res. 2009, 15, 2913–2919. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef]
- Ceppi, P.; Volante, M.; Novello, S.; Rapa, I.; Danenberg, K.D.; Danenberg, P.V.; Cambieri, A.; Selvaggi, G.; Saviozzi, S.; Calogero, R.; et al. ERCC1 and RRM1 gene expressions but not EGFR are predictive of shorter survival in advanced non-small-cell lung cancer treated with cisplatin and gemcitabine. Ann. Oncol. 2006, 17, 1818–1825. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Clarke, M.L.; Falette, N.; Puisieux, A.; Mackey, J.R.; Dumontet, C. Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int. J. Cancer 2002, 97, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-W.; Liu, J.-Y.; Lin, S.-T.; Li, J.-M.; Huang, S.-H.; Chen, J.-Y.; Wu, J.-Y.; Kuo, C.-C.; Wu, C.-L.; Lu, Y.-C.; et al. Proteomic analysis of gemcitabine-induced drug resistance in pancreatic cancer cells. Mol. Biosyst. 2011, 7, 3065–3074. [Google Scholar] [CrossRef]
- Samulitis, B.K.; Pond, K.W.; Pond, E.; Cress, A.E.; Patel, H.; Wisner, L.; Patel, C.; Dorr, R.T.; Landowski, T.H. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol. Ther. 2015, 16, 43–51. [Google Scholar] [CrossRef]
- Fujimura, Y.; Ikenaga, N.; Ohuchida, K.; Setoyama, D.; Irie, M.; Miura, D.; Wariishi, H.; Murata, M.; Mizumoto, K.; Hashizume, M.; et al. Mass spectrometry-based metabolic profiling of gemcitabine-sensitive and gemcitabine-resistant pancreatic cancer cells. Pancreas 2014, 43, 311–318. [Google Scholar] [CrossRef]
- Lagies, S.; Pichler, R.; Kaminski, M.M.; Schlimpert, M.; Walz, G.; Lienkamp, S.S.; Kammerer, B. Metabolic characterization of directly reprogrammed renal tubular epithelial cells (iRECs). Sci. Rep. 2018, 8, 3878. [Google Scholar] [CrossRef] [Green Version]
- SpectConnect: Analysis of GC/MS Datasets. Available online: http://spectconnect.mit.edu/ (accessed on 27 October 2019).
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Yunis, A.A.; Arimura, G.K.; Russin, D.J. Human pancreatic carcinoma (MIA PaCa-2) in continuous culture: Sensitivity to asparaginase. Int. J. Cancer 1977, 19, 128–135. [Google Scholar] [CrossRef]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.A.; Galanis, C.; Woo, Y.; Chen, N.; Zhang, Q.; Fong, Y.; Szalay, A.A. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1h68. Mol. Cancer Ther. 2009, 8, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagies, S.; Pichler, R.; Bork, T.; Kaminski, M.M.; Troendle, K.; Zimmermann, S.; Huber, T.B.; Walz, G.; Lienkamp, S.S.; Kammerer, B. Impact of Diabetic Stress Conditions on Renal Cell Metabolome. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Siddiqui, A.; Vazakidou, M.E.; Napoli, F.; Böttcher, M.; Menchicchi, B.; Raza, U.; Saatci, Ö.; Krebs, A.M.; Ferrazzi, F.; et al. Polyol Pathway Links Glucose Metabolism to the Aggressiveness of Cancer Cells. Cancer Res. 2018, 78, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—Pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Ripps, H.; Shen, W. Review: Taurine: A “very essential” amino acid. Mol. Vis. 2012, 18, 2673–2686. [Google Scholar]
- White-Gilbertson, S.; Kurtz, D.T.; Voelkel-Johnson, C. The role of protein synthesis in cell cycling and cancer. Mol. Oncol. 2009, 3, 402–408. [Google Scholar] [CrossRef]
- Thomas, T.; Thomas, T.J. Polyamines in cell growth and cell death: Molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. CMLS 2001, 58, 244–258. [Google Scholar] [CrossRef]
- Tahiliani, A.G.; Beinlich, C.J. Pantothenic acid in health and disease. Vitam. Horm. 1991, 46, 165–228. [Google Scholar]
- Szende, B. The effect of amino acids and amino acid derivatives on cell proliferation. Acta Bio-Med. Ateneo Parm. Organo Della Soc. Med. E Sci. Nat. Parma 1993, 64, 139–145. [Google Scholar]
- Copley, S.D.; Frank, E.; Kirsch, W.M.; Koch, T.H. Detection and possible origins of aminomalonic acid in protein hydrolysates. Anal. Biochem. 1992, 201, 152–157. [Google Scholar] [CrossRef]
- Ng, B.G.; Wolfe, L.A.; Ichikawa, M.; Markello, T.; He, M.; Tifft, C.J.; Gahl, W.A.; Freeze, H.H. Biallelic mutations in CAD, impair de novo pyrimidine biosynthesis and decrease glycosylation precursors. Hum. Mol. Genet. 2015, 24, 3050–3057. [Google Scholar] [CrossRef] [Green Version]
- Rao, T.V.P.; Kuzminov, A. Sources of thymidine and analogs fueling futile damage-repair cycles and ss-gap accumulation during thymine starvation in Escherichia coli. DNA Repair 2019, 75, 1–17. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Wada, Y.; Endo, S.; Soda, M.; El-Kabbani, O.; Hara, A. Aldo–Keto Reductase 1B10 and Its Role in Proliferation Capacity of Drug-Resistant Cancers. Front. Pharmacol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Sandforth, L.; Ammar, N.; Dinges, L.A.; Röcken, C.; Arlt, A.; Sebens, S.; Schäfer, H. Impact of the Monocarboxylate Transporter-1 (MCT1)-Mediated Cellular Import of Lactate on Stemness Properties of Human Pancreatic Adenocarcinoma Cells. Cancers 2020, 12, 581. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, M.M.A.; Neidigh, J.W.; Wall, N.R. Antimetabolite Treatment for Pancreatic Cancer. Chemotherapy 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399. [Google Scholar] [CrossRef]
- Brown, K.K.; Spinelli, J.B.; Asara, J.; Toker, A. Adaptive Reprogramming of De Novo Pyrimidine Synthesis is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discov. 2017, 7, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Swyryd, E.A.; Seaver, S.S.; Stark, G.R. N-(Phosphonacetyl)-l-Aspartate, a Potent Transition State Analog Inhibitor of Aspartate Transcarbamylase, Blocks Proliferation of Mammalian Cells in Culture. J. Biol. Chem. 1974, 249, 6945–6950. [Google Scholar]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Parental IC50 | Resistant IC50 | Fold Change |
|---|---|---|---|
| MiaPaCa-2 | 0.0041 nM | 0.31 nM | 74.7 |
| Panc-1 | 0.0073 nM | 0.25 nM | 33.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, L.M.; Lagies, S.; Guenzle, J.; Fichtner-Feigl, S.; Wittel, U.A.; Kammerer, B. Metabolic Adaptation during nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines. Cells 2020, 9, 1251. https://doi.org/10.3390/cells9051251
Braun LM, Lagies S, Guenzle J, Fichtner-Feigl S, Wittel UA, Kammerer B. Metabolic Adaptation during nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines. Cells. 2020; 9(5):1251. https://doi.org/10.3390/cells9051251
Chicago/Turabian StyleBraun, Lukas M., Simon Lagies, Jessica Guenzle, Stefan Fichtner-Feigl, Uwe A. Wittel, and Bernd Kammerer. 2020. "Metabolic Adaptation during nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines" Cells 9, no. 5: 1251. https://doi.org/10.3390/cells9051251
APA StyleBraun, L. M., Lagies, S., Guenzle, J., Fichtner-Feigl, S., Wittel, U. A., & Kammerer, B. (2020). Metabolic Adaptation during nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines. Cells, 9(5), 1251. https://doi.org/10.3390/cells9051251