Effects of β-Adrenergic Blockade on Metabolic and Inflammatory Responses in a Rat Model of Ischemic Stroke

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
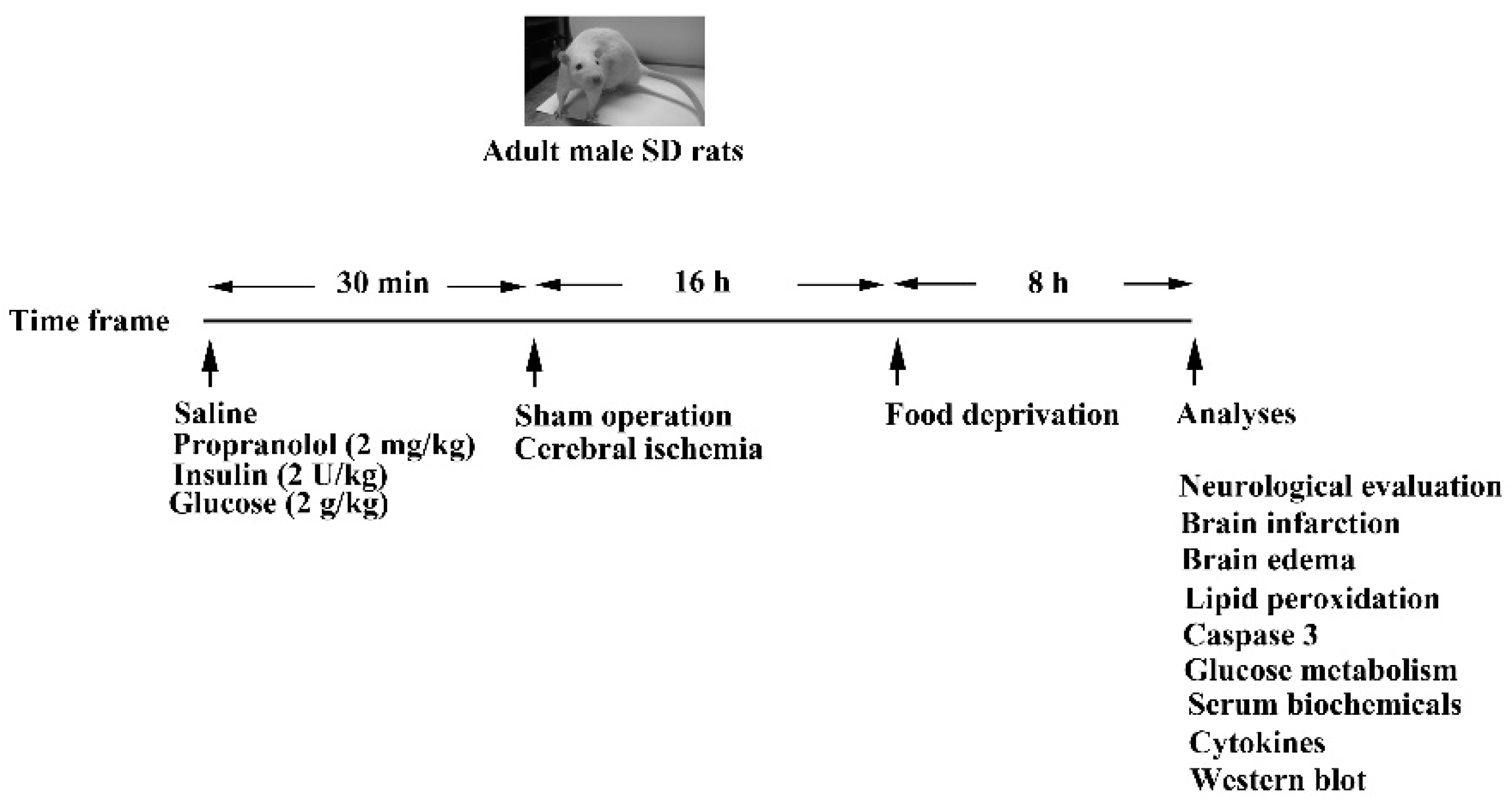
2.1. Cerebral Ischemia and Treatments
2.2. Neurological Evaluation
2.3. Quantification of Ischemic Infarction
2.4. Brain Edema
2.5. Measurement of Lipid Peroxidation
2.6. Caspase 3 Activity Assay
2.7. Glucose Tolerance Test
2.8. Blood Sample Analyses
2.9. Measurement of Tissue Cytokines
2.10. Western Blot Analysis
2.11. Cell Cultures
2.12. Statistical Analysis
3. Results
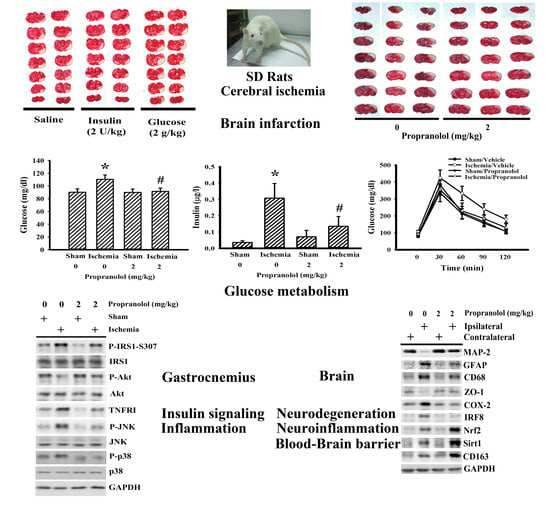
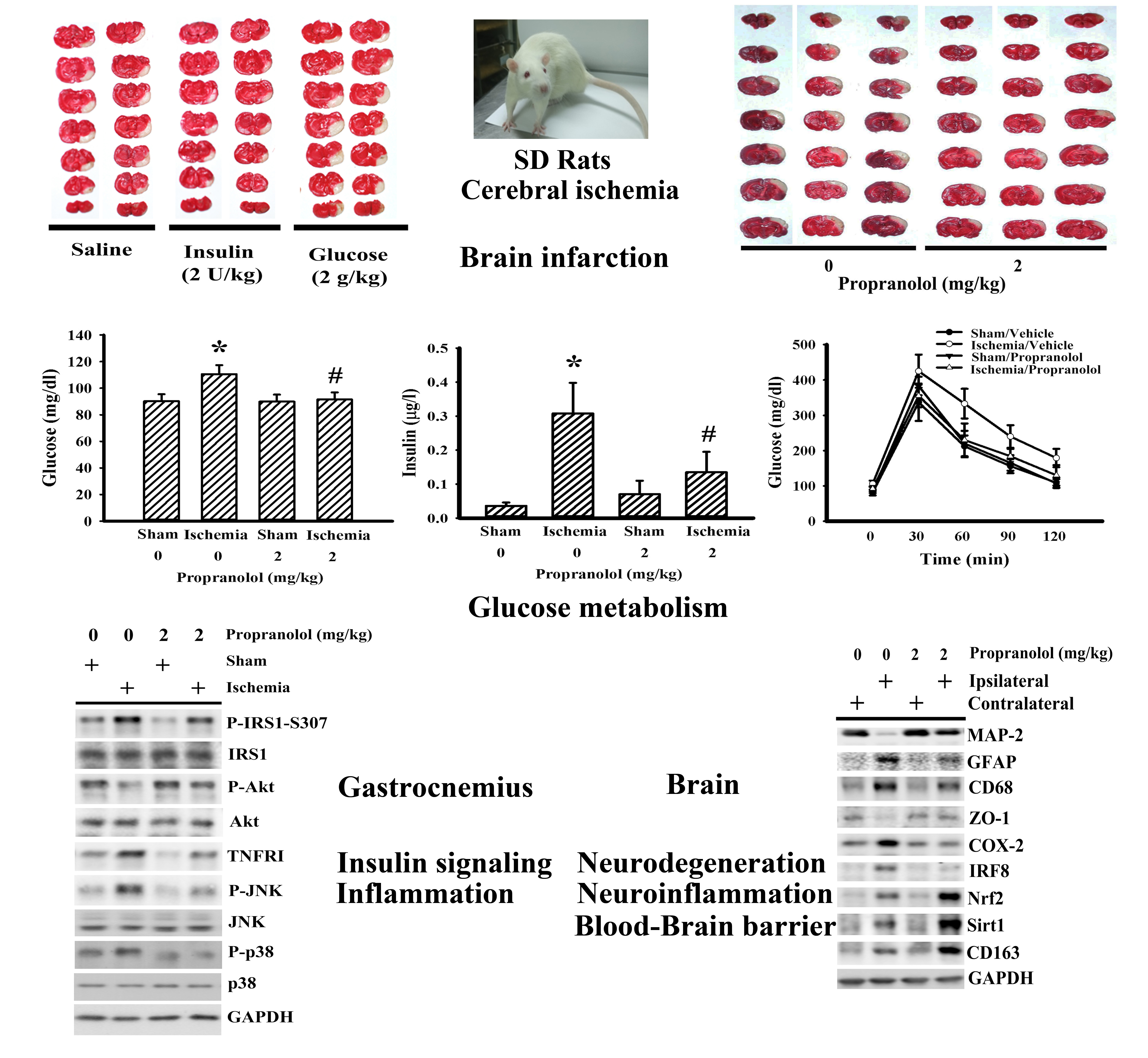
3.1. Propranolol Alleviated Postischemic Brain Injury
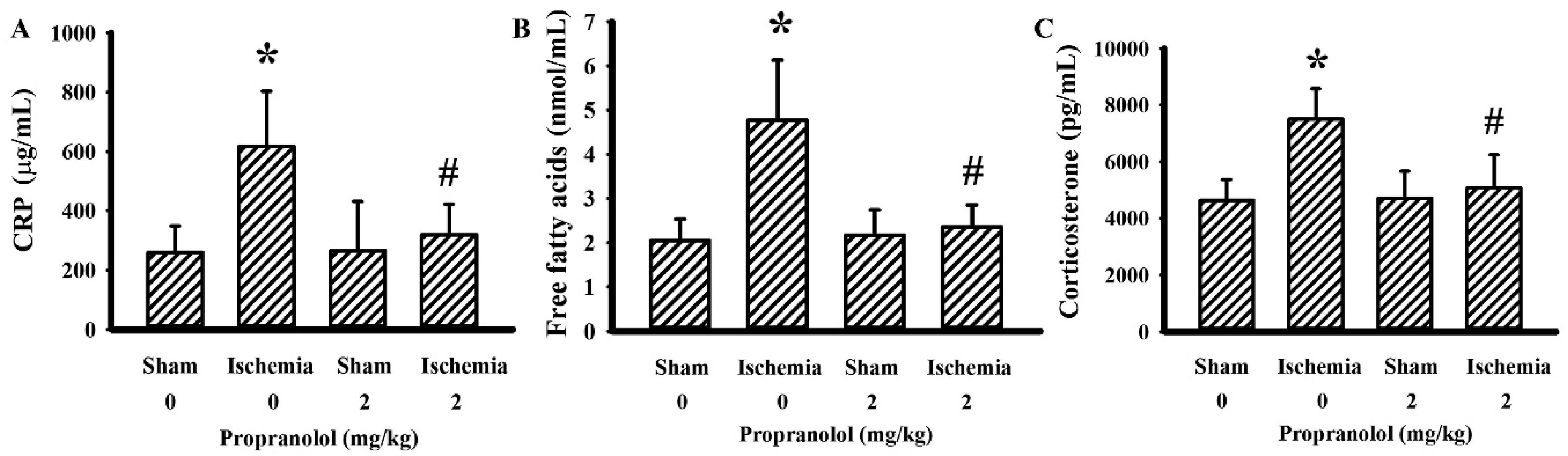
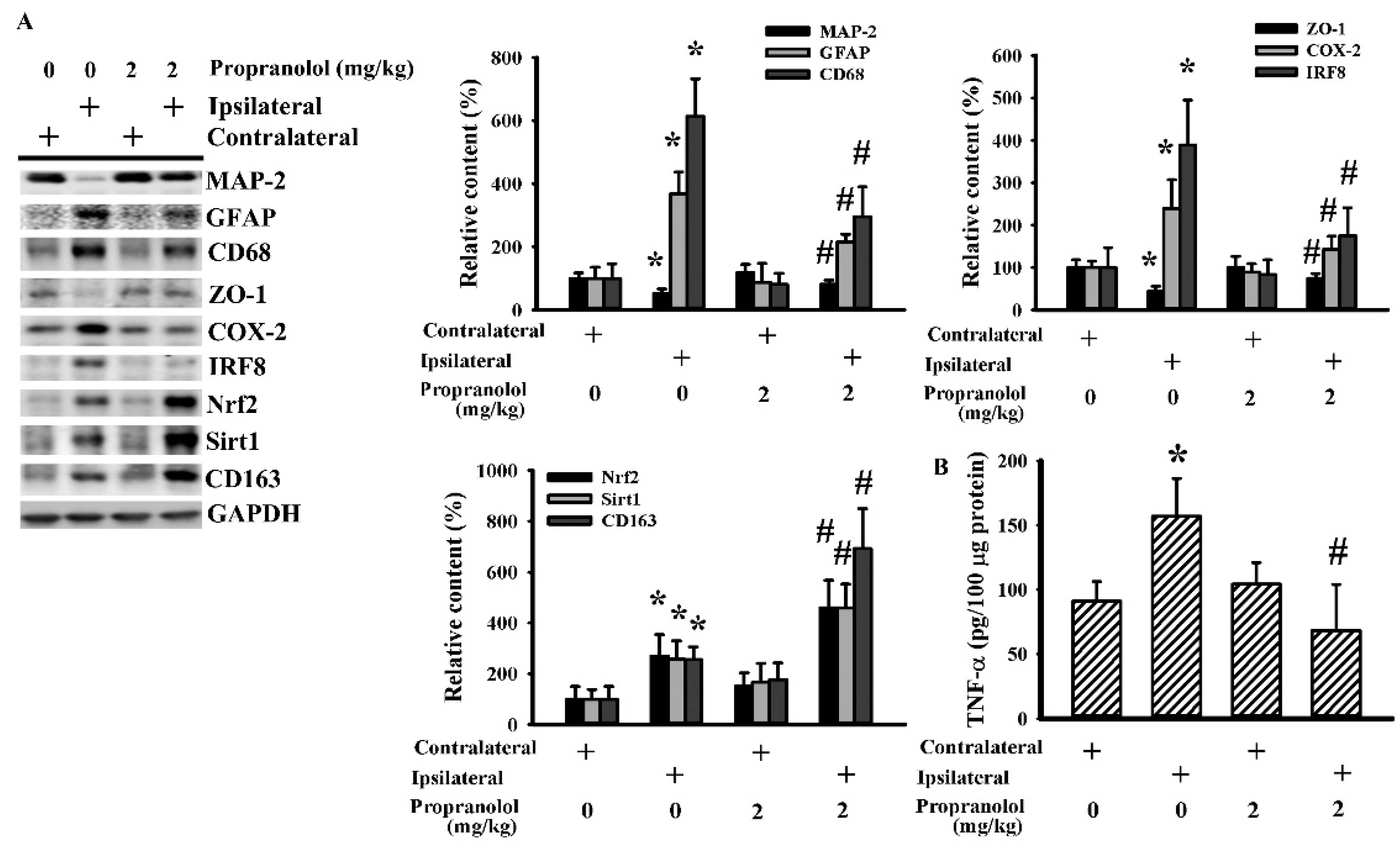
3.2. Propranolol Alleviated Postischemic Inflammation
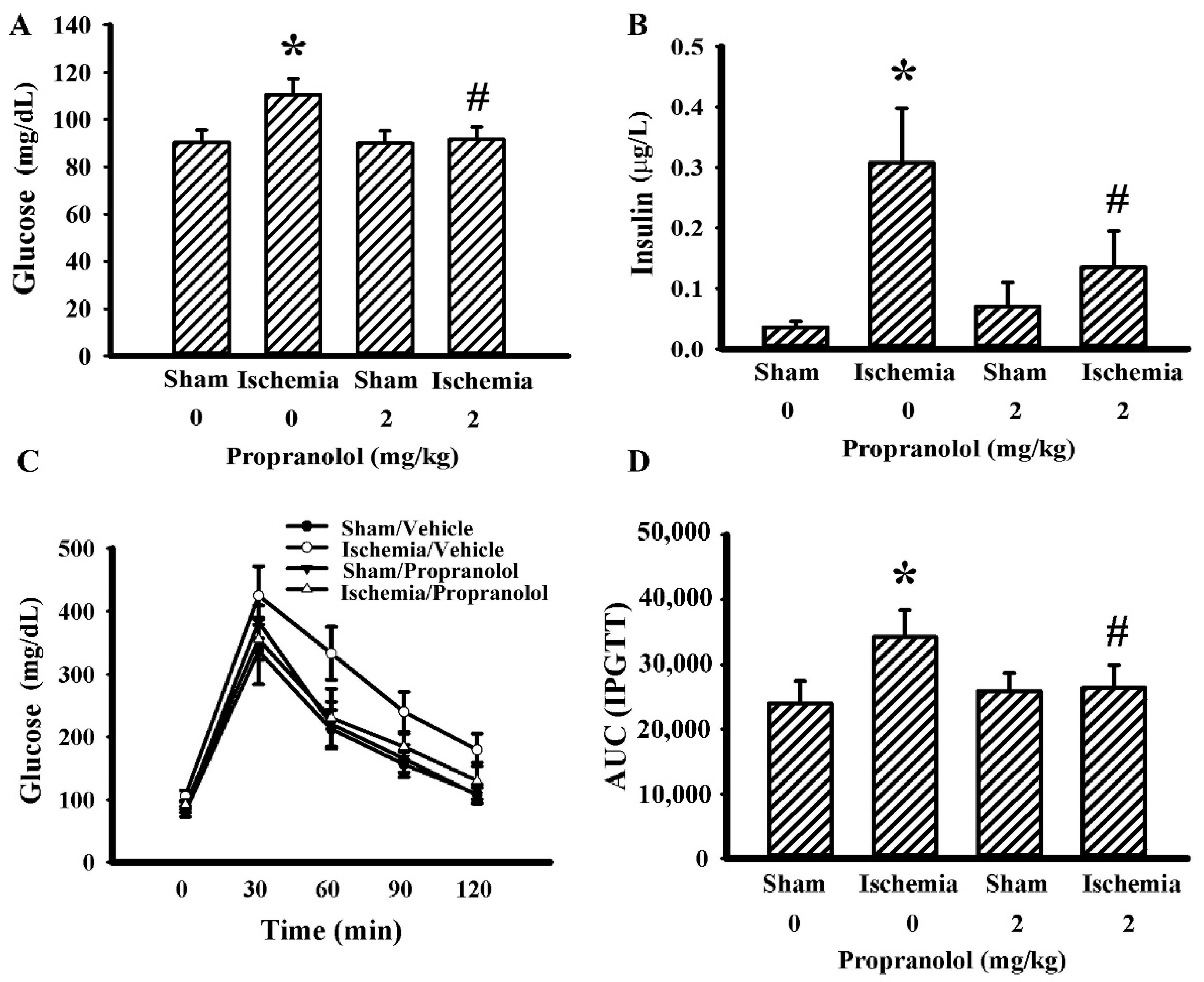
3.3. Propranolol Improved Postischemic Hyperglycemia
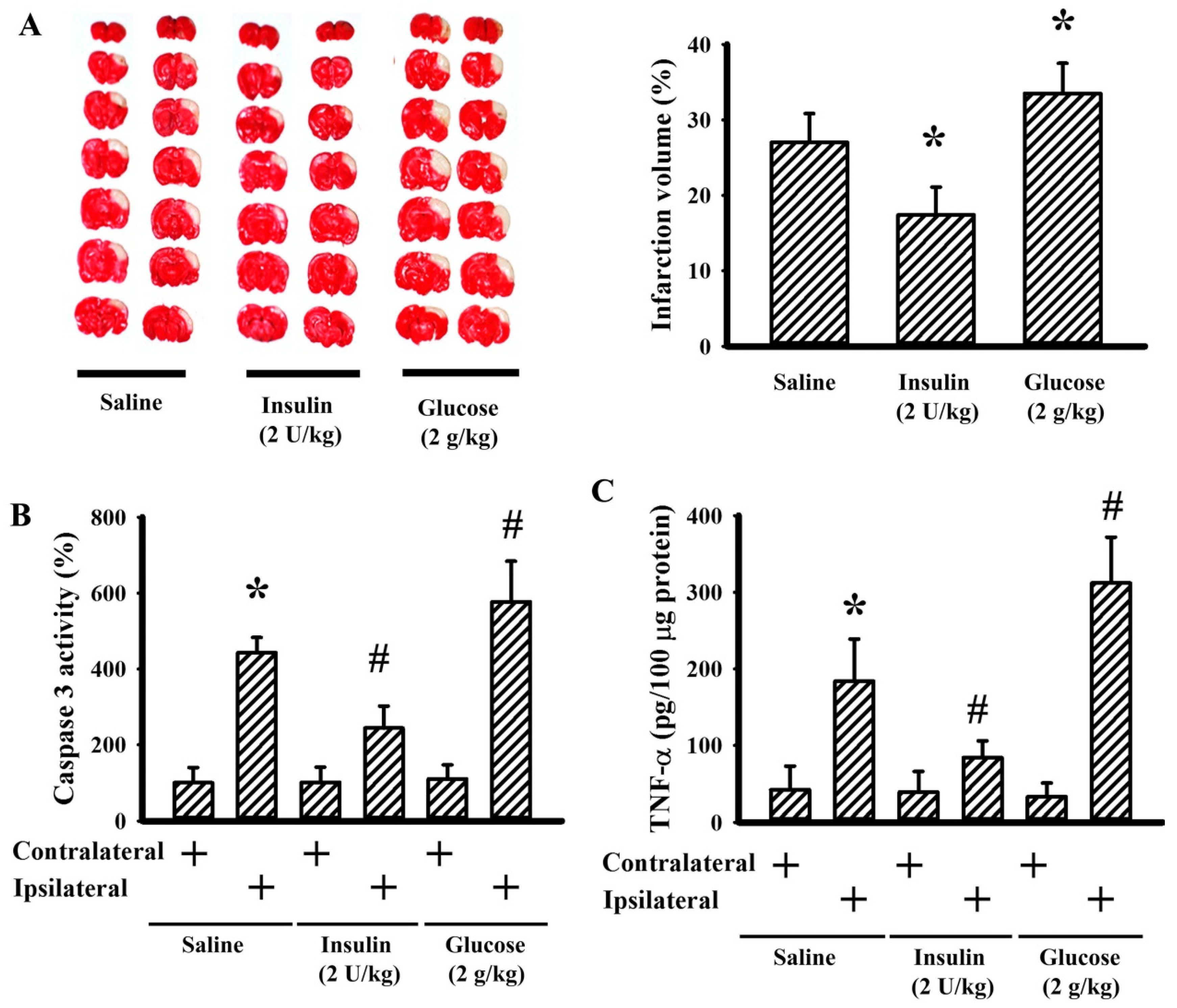
3.4. Insulin and Glucose Had Opposite Effects on Postischemic Changes
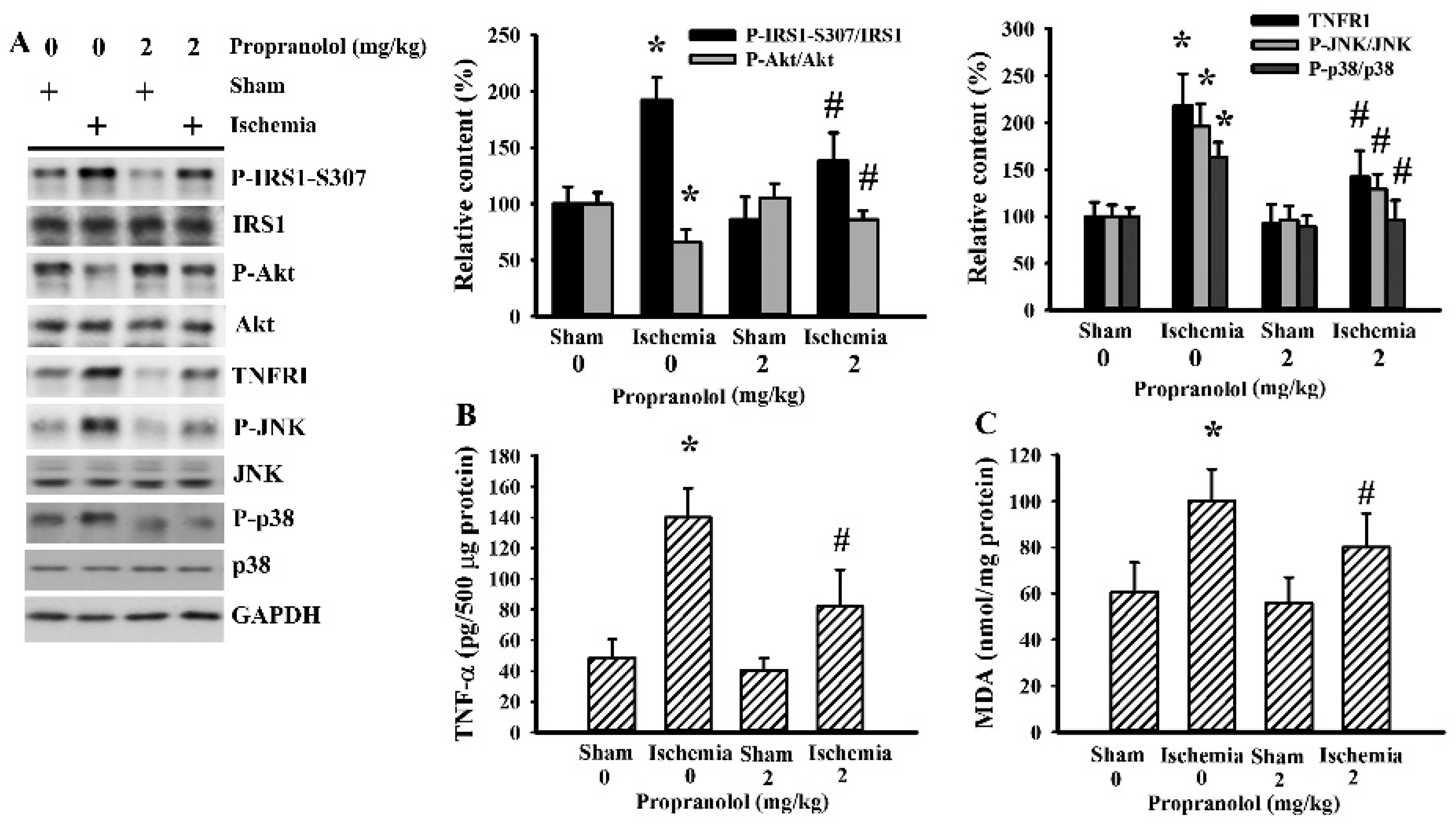
3.5. Cerebral Ischemia Impaired Insulin Action in Gastrocnemius
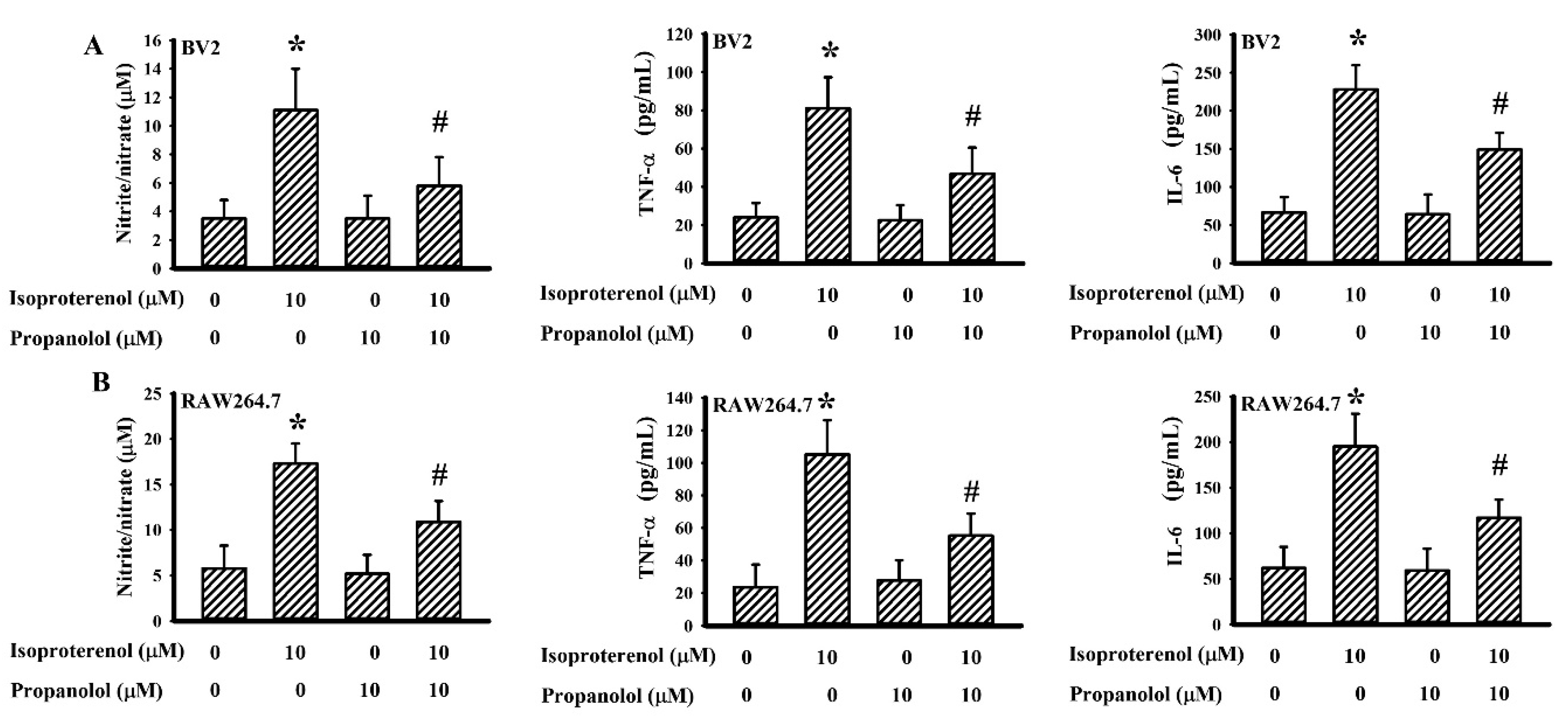
3.6. Propranolol Decreased Isoproterenol-Induced Cytokine Production
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Boehme, A.K.; Kapoor, N.; Albright, K.C.; Lyerly, M.J.; Rawal, P.V.; Bavarsad Shahripour, R.; Alvi, M.; Houston, J.T.; Sisson, A.; Beasley, T.M.; et al. Predictors of systemic inflammatory response syndrome in ischemic stroke undergoing systemic thrombolysis with intravenous tissue plasminogen activator. J. Stroke Cerebrovasc. Dis. 2014, 23, e271–e276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafez, S.; Coucha, M.; Bruno, A.; Fagan, S.C.; Ergul, A. Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Transl. Stroke Res. 2014, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, M.; Takahashi, T.; Nishizawa, M.; Shimohata, T. Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. J. Atheroscler. Thromb. 2017, 24, 240–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero-Ortega, L.; Gutiérrez-Fernández, M.; Gutiérrez-Zúñiga, R.; Madero-Jarabo, R.; de Leciñana, M.A.; Laso-García, F.; Lisbona, A.; Delgado-Mederos, R.; Gállego-Culleré, J.; Martínez-Zabaleta, M.; et al. The effect of post-stroke hyperglycaemia on the levels of brain damage and repair-related circulating biomarkers: The Glycaemia in Acute Stroke Study II. Eur. J. Neurol. 2019, 26, 1439–1446. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, J.; Zhang, J.; Xu, T.; Zhang, H.; Zhang, Y.; Zhang, S. Association of stroke clinical outcomes with coexistence of hyperglycemia and biomarkers of inflammation. J. Stroke Cerebrovasc. Dis. 2015, 24, 1250–1255. [Google Scholar] [CrossRef]
- Chen, L.; Cao, J.; Cao, D.; Wang, M.; Xiang, H.; Yang, Y.; Ying, T.; Cong, H. Protective effect of dexmedetomidine against diabetic hyperglycemia-exacerbated cerebral ischemia/reperfusion injury: An in vivo and in vitro study. Life Sci. 2019, 235, 116553. [Google Scholar] [CrossRef]
- Desilles, J.P.; Syvannarath, V.; Ollivier, V.; Journé, C.; Delbosc, S.; Ducroux, C.; Boisseau, W.; Louedec, L.; Di Meglio, L.; Loyau, S.; et al. Exacerbation of thromboinflammation by hyperglycemia precipitates cerebral infarct growth and hemorrhagic transformation. Stroke 2017, 48, 1932–1940. [Google Scholar] [CrossRef]
- Ismael, S.; Nasoohim, S.; Yoo, A.; Ahmed, H.A.; Ishrat, T. Tissue plasminogen activator promotes TXNIP-NLRP3 inflammasome activation after hyperglycemic stroke in mice. Mol. Neurobiol. 2020, 57, 2495–2508. [Google Scholar] [CrossRef]
- Khan, M.A.; Schultz, S.; Othman, A.; Fleming, T.; Lebrón-Galán, R.; Rades, D.; Clemente, D.; Nawroth, P.P.; Schwaninger, M. Hyperglycemia in stroke impairs polarization of monocytes/macrophages to a protective noninflammatory cell type. J. Neurosci. 2016, 36, 9313–9325. [Google Scholar] [CrossRef] [Green Version]
- Won, S.J.; Tang, X.N.; Suh, S.W.; Yenari, M.A.; Swanson, R.A. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann. Neurol. 2011, 70, 583–590. [Google Scholar] [CrossRef]
- Scanzano, A.; Cosentino, M. Adrenergic regulation of innate immunity: A review. Front. Pharmacol. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Chen, Y.; Li, J.; Liu, Z.; Wang, Z.; Chen, J.; Cao, W.; Xu, Y. Cocaine-and amphetamine-regulated transcript modulates peripheral immunity and protects against brain injury in experimental stroke. Brain Behav. Immun. 2011, 25, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Radak, D.; Resanovic, I.; Isenovic, E.R. Changes in hypothalamus-pituitary-adrenal axis following transient ischemic attack. Angiology 2014, 65, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chen, C.J.; Lin, S.Y.; Chuang, Y.H.; Sheu, W.H.; Tung, K.C. Hyperglycemia is associated with enhanced gluconeogenesis in a rat model of permanent cerebral ischemia. Mol. Cell. Endocrinol. 2013, 367, 50–56. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Lin, S.Y.; Chuang, Y.H.; Chen, C.J.; Tung, K.C.; Sheu, W.H. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E155–E163. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Lin, S.Y.; Chuang, Y.H.; Sheu, W.H.; Tung, K.C.; Chen, C.J. Activation of hepatic inflammatory pathways by catecholamines is associated with hepatic insulin resistance in male ischemic stroke rats. Endocrinology 2014, 155, 1235–1246. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.L.; Zhang, J.H. Role of the sympathetic nervous system and spleen in experimental stroke-induced immunodepression. Med. Sci. Monit. 2014, 20, 2489–2496. [Google Scholar]
- Zuo, L.; Shi, L.; Yan, F. The reciprocal interaction of sympathetic nervous system and cAMP-PKA-NF-kB pathway in immune suppression after experimental stroke. Neurosci. Lett. 2016, 627, 205–210. [Google Scholar] [CrossRef]
- Lechtenberg, K.J.; Meyer, S.T.; Doyle, J.B.; Peterson, T.C.; Buckwalter, M.S. Augmented β2-adrenergic signaling dampens the neuroinflammatory response following ischemic stroke and increases stroke size. J. Neuroinflammation 2019, 16, 112. [Google Scholar] [CrossRef] [Green Version]
- Goyagi, T.; Kimura, T.; Nishikawa, T.; Tobe, Y.; Masaki, Y. Beta-adrenoreceptor antagonists attenuate brain injury after transient focal ischemia in rats. Anesth. Analg. 2006, 103, 658–663. [Google Scholar] [CrossRef]
- Iwata, M.; Inoue, S.; Kawaguchi, M.; Nakamura, M.; Konishi, N.; Furuya, H. Posttreatment but not pretreatment with selective beta-adrenoreceptor 1 antagonists provides neuroprotection in the hippocampus in rats subjected to transient forebrain ischemia. Anesth. Analg. 2010, 110, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Luger, S.; Schwebler, A.; Vutukuri, R.; Bouzas, N.F.; Labocha, S.; Schreiber, Y.; Brunkhorst, R.; Steinmetz, H.; Pfeilschifter, J.; Pfeilschifter, W. Beta adrenoceptor blockade ameliorates impaired glucose tolerance and alterations of the cerebral ceramide metabolism in an experimental model of ischemic stroke. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.D.; Zimomra, Z.R.; Stewart, L.T. Beta-adrenergic receptor activation primes microglia cytokine production. J. Neuroimmunol. 2013, 254, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ruan, L.; Qian, L.; Liu, X.; Le, Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci. 2010, 30, 11848–11857. [Google Scholar] [CrossRef] [Green Version]
- Tozaki-Saitoh, H.; Sasaki, I.; Yamashita, T.; Hosoi, M.; Kato, T.A.; Tsuda, M. Involvement of exchange protein directly activated by cAMP and tumor progression locus 2 in IL-1b production in microglial cells following activation of b-adrenergic receptors. J. Pharmacol. Sci. 2020. [CrossRef]
- Wang, J.; Li, J.; Sheng, X.; Zhao, H.; Cao, X.D.; Wang, Y.Q.; Wu, G.C. Beta-adrenoceptor mediated surgery-induced production of pro-inflammatory cytokines in rat microglia cells. J. Neuroimmunol. 2010, 223, 77–83. [Google Scholar] [CrossRef]
- Chang, C.Y.; Kuan, Y.H.; Li, J.R.; Chen, W.Y.; Ou, Y.C.; Pan, H.C.; Liao, S.L.; Raung, S.L.; Chang, C.J.; Chen, C.J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef]
- Chen, W.Y.; Mao, F.C.; Liu, C.H.; Kuan, Y.H.; Lai, N.W.; Wu, C.C.; Chen, C.J. Chromium supplementation improved post-stroke brain infarction and hyperglycemia. Metab. Brain Dis. 2016, 31, 289–297. [Google Scholar] [CrossRef]
- Webb, A.J.; Fischer, U.; Rothwell, P.M. Effects of β-blocker selectivity on blood pressure variability and stroke: A systematic review. Neurology 2011, 77, 731–737. [Google Scholar] [CrossRef]
- Chen, C.; Raung, S.L.; Liao, S.L.; Chen, S.Y. Inhibition of inducible nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimulated microglia. Biochem. Pharmacol. 2004, 67, 957–965. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chen, C.J.; Liu, C.H.; Mao, C.F. Chromium supplementation enhances insulin signaling in skeletal muscle of obese KK/HlJ diabetic mice. Diabetes Obes. Metab. 2009, 11, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Cerecedo-Lopez, C.D.; Cantu-Aldana, A.; Patel, N.J.; Aziz-Sultan, M.A.; Frerichs, K.U.; Du, R. Insulin in the management of acute ischemic stroke: A systematic review and meta-analysis. World Neurosurg. 2020, 136, e514–e534. [Google Scholar] [CrossRef]
- Fan, X.; Ning, M.; Lo, E.H.; Wang, X. Early insulin glycemic control combined with tPA thrombolysis reduces acute brain tissue damages in a focal embolic stroke model of diabetic rats. Stroke 2013, 44, 255–259. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, L.; Lu, M.; Hu, W.; Xiu, S. Hyperglycemia is associated with poor in-hospital outcome in elderly patients with acute ischemic stroke. Medicine (Baltimore) 2019, 98, e16723. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, K.; Usui, I.; Kanatani, Y.; Bukhari, A.; He, J.; Fujisaka, S.; Yamazaki, Y.; Suzuki, H.; Hiratani, K.; Ishiki, M.; et al. Chronic tumor necrosis factor-alpha treatment causes insulin resistance via insulin receptor substrate-1 serine phosphorylation and suppressor of cytokine signaling-3 induction in 3T3-L1 adipocytes. Endocrinology 2007, 148, 2994–3003. [Google Scholar] [CrossRef]
- Long, M.H.; Zhang, C.; Xu, D.Q.; Fu, W.L.; Gan, X.D.; Li, F.; Wang, Q.; Xia, W.; Xu, D.G. PM2.5 aggravates diabetes via the systemically activated IL-6-mediated STAT3/SOCS3 pathway in rats’ liver. Environ. Pollut. 2020, 256, 113342. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yu, H.; Xiao, Y.; Wang, H. Momordica charantia extracts ameliorate insulin resistance by regulating the expression of SOCS-3 and JNK in type 2 diabetes mellitus rats. Pharm. Biol. 2017, 55, 2170–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hartung, J.E.; Bortsov, A.V.; Kim, S.; O’Buckley, S.C.; Kozlowski, J.; Nackley, A.G. Sustained stimulation of β2- and β3-adrenergic receptors leads to persistent functional pain and neuroinflammation. Brain Behav. Immun. 2018, 73, 520–532. [Google Scholar] [CrossRef]
- Chen, C.; Du, J.; Feng, W.; Song, Y.; Lu, Z.; Xu, M.; Li, Z.; Zhang, Y. β-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCd/p38 MAPK signaling in neonatal mouse cardiac fibroblasts. Br. J. Pharmacol. 2012, 166, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Kolmus, K.; Van Troys, M.; Van Wesemael, K.; Ampe, C.; Haegeman, G.; Tavernier, J.; Gerlo, S. b-agonists selectively modulate proinflammatory gene expression in skeletal muscle cells via non-canonical nuclear crosstalk mechanisms. PLoS ONE 2014, 9, e90649. [Google Scholar] [CrossRef]
- Armstead, W.M.; Vavilala, M.S. Propranolol protects cerebral autoregulation and reduces hippocampal neuronal cell death through inhibition of interleukin-6 upregulation after traumatic brain injury in pigs. Br. J. Anaesth. 2019, 123, 610–617. [Google Scholar] [CrossRef] [PubMed]
- García-Miss Mdel, R.; Mut-Martín, M.C.; Góngora-Alfaro, J.L. b-Adrenergic blockade protects BALB/c mice against infection with a small inoculum of Leishmania mexicana mexicana (LV4). Int. Immunopharmacol. 2015, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Özyılmaz, E.; Büyüknacar, H.S.G.; Bağır, E.K.; Sencar, L.; Öztürk, Ö.G.; Eray, I.C.; Dağlıoğlu, Y.K.; Baydar, O.; Seydaoğlu, G.; Mete, U.Ö.; et al. Early propranolol treatment ameliorates endothelial dysfunction in experimental septic lung. Adv. Clin. Exp. Med. 2019, 28, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Jin, T.; Zhang, X.; Zeng, Z.; Ye, B.; Wang, J.; Zhong, Y.; Xiong, X.; Gu, L. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-κB signaling pathway. Front. Cell. Neurosci. 2019, 13, 553. [Google Scholar] [CrossRef] [Green Version]
- Al Mamun, A.; Chauhan, A.; Yu, H.; Xu, Y.; Sharmeen, R.; Liu, F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur. J. Neurosci. 2018, 47, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Bok, E.; Chung, Y.C.; Kim, K.S.; Baik, H.H.; Shin, W.H.; Jin, B.K. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Exp. Mol. Med. 2018, 50, 76. [Google Scholar] [CrossRef] [Green Version]
- Ślusarczyk, J.; Trojan, E.; Głombik, K.; Piotrowska, A.; Budziszewska, B.; Kubera, M.; Popiołek-Barczyk, K.; Lasoń, W.; Mika, J.; Basta-Kaim, A. Targeting the NLRP3 Inflammasome-related pathways via tianeptine treatment-suppressed microglia polarization to the M1 phenotype in lipopolysaccharide-stimulated cultures. Int. J. Mol. Sci. 2018, 19, 1965. [Google Scholar] [CrossRef] [Green Version]
- Velagapudi, R.; Jamshaid, F.; Lepiarz, I.; Katola, F.O.; Hemming, K.; Olajide, O.A. The tiliroside derivative, 3-O-[(E)-(2-oxo-4-(p-tolyl) but-3-en-1-yl] kaempferol produced inhibition of neuroinflammation and activation of AMPK and Nrf2/HO-1 pathways in BV-2 microglia. Int. Immunopharmacol. 2019, 77, 105951. [Google Scholar] [CrossRef]
- Dasu, M.R.; Ramirez, S.R.; La, T.D.; Gorouhi, F.; Nguyen, C.; Lin, B.R.; Mashburn, C.; Stewart, H.; Peavy, T.R.; Nolta, J.A.; et al. Crosstalk between adrenergic and toll-like receptors in human mesenchymal stem cells and keratinocytes: A recipe for impaired wound healing. Stem Cells Transl. Med. 2014, 3, 745–759. [Google Scholar] [CrossRef]
- Zhou, J.; Yan, J.; Liang, H.; Jiang, J. Epinephrine enhances the response of macrophages under LPS stimulation. Biomed. Res. Int. 2014, 2014, 254686. [Google Scholar] [CrossRef] [Green Version]
- Grailer, J.J.; Haggadone, M.D.; Sarma, J.V.; Zetoune, F.S.; Ward, P.A. Induction of M2 regulatory macrophages through the b2-adrenergic receptor with protection during endotoxemia and acute lung injury. J. Innate Immun. 2014, 6, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, T.; Pera, J.; Zur-Wyrozumska, K.; Klimkowicz-Mrowiec, A.; Szczudlik, A.; Slowik, A. Beta-blockers use and risk of hyperglycemia in acute stroke patients. Atherosclerosis 2012, 223, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Monai, H.; Wang, X.; Yahagi, K.; Lou, N.; Mestre, H.; Xu, Q.; Abe, Y.; Yasui, M.; Iwai, Y.; Nedergaard, M.; et al. Adrenergic receptor antagonism induces neuroprotection and facilitates recovery from acute ischemic stroke. Proc. Natl. Acad. Sci. USA 2019, 116, 11010–11019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.R.; Ou, Y.C.; Wu, C.C.; Wang, J.D.; Lin, S.Y.; Wang, Y.Y.; Chen, W.Y.; Chen, C.J. Ischemic preconditioning improved renal ischemia/reperfusion injury and hyperglycemia. IUBMB Life. 2019, 71, 321–329. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-Y.; Wang, Y.-Y.; Chang, C.-Y.; Wu, C.-C.; Chen, W.-Y.; Kuan, Y.-H.; Liao, S.-L.; Chen, C.-J. Effects of β-Adrenergic Blockade on Metabolic and Inflammatory Responses in a Rat Model of Ischemic Stroke. Cells 2020, 9, 1373. https://doi.org/10.3390/cells9061373
Lin S-Y, Wang Y-Y, Chang C-Y, Wu C-C, Chen W-Y, Kuan Y-H, Liao S-L, Chen C-J. Effects of β-Adrenergic Blockade on Metabolic and Inflammatory Responses in a Rat Model of Ischemic Stroke. Cells. 2020; 9(6):1373. https://doi.org/10.3390/cells9061373
Chicago/Turabian StyleLin, Shih-Yi, Ya-Yu Wang, Cheng-Yi Chang, Chih-Cheng Wu, Wen-Ying Chen, Yu-Hsiang Kuan, Su-Lan Liao, and Chun-Jung Chen. 2020. "Effects of β-Adrenergic Blockade on Metabolic and Inflammatory Responses in a Rat Model of Ischemic Stroke" Cells 9, no. 6: 1373. https://doi.org/10.3390/cells9061373
APA StyleLin, S. -Y., Wang, Y. -Y., Chang, C. -Y., Wu, C. -C., Chen, W. -Y., Kuan, Y. -H., Liao, S. -L., & Chen, C. -J. (2020). Effects of β-Adrenergic Blockade on Metabolic and Inflammatory Responses in a Rat Model of Ischemic Stroke. Cells, 9(6), 1373. https://doi.org/10.3390/cells9061373