The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment

 and
and 
Abstract
:1. Introduction
2. GSK-3 Inhibitors
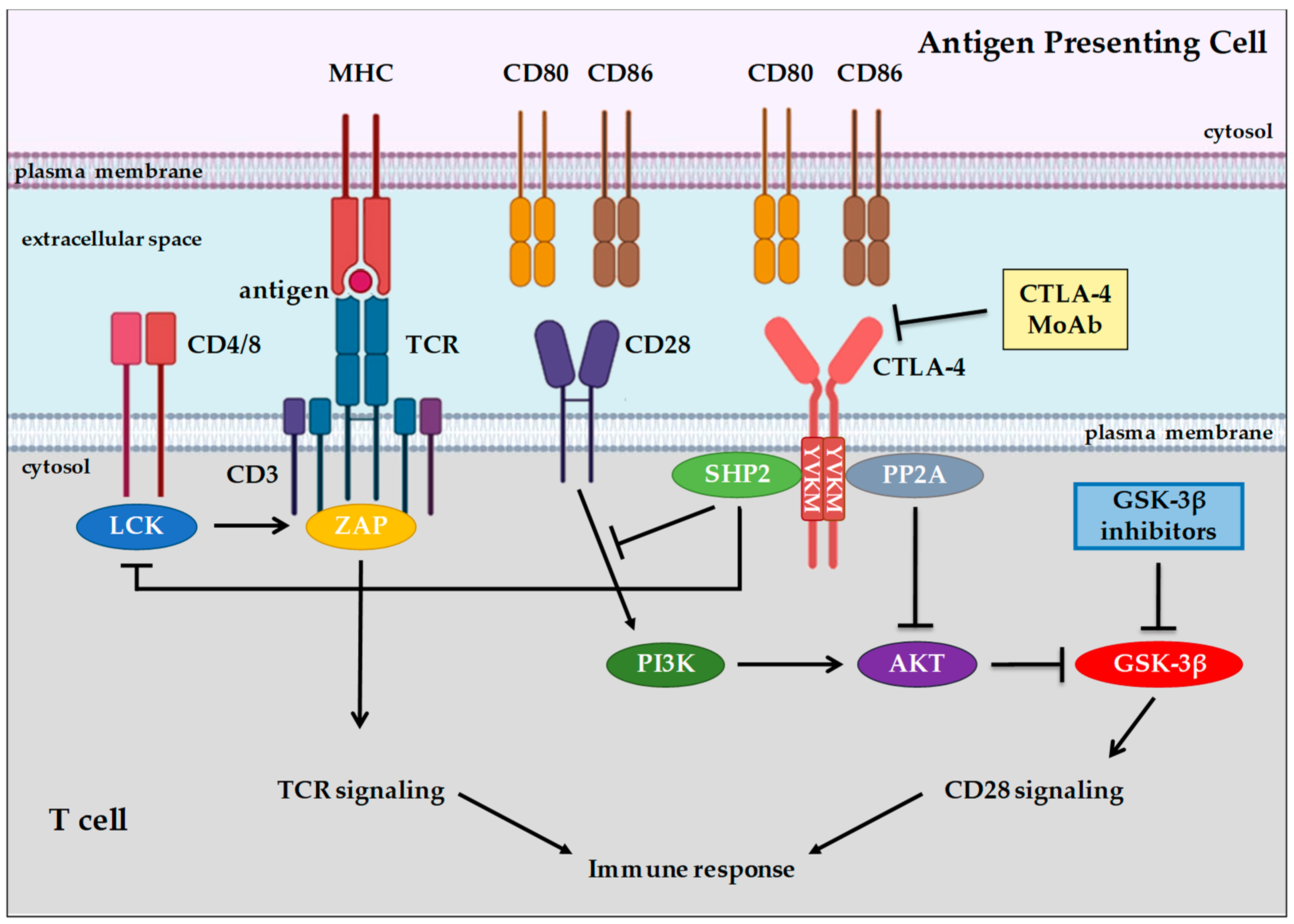
3. Background of Immune System Cells and Immune Checkpoint Proteins
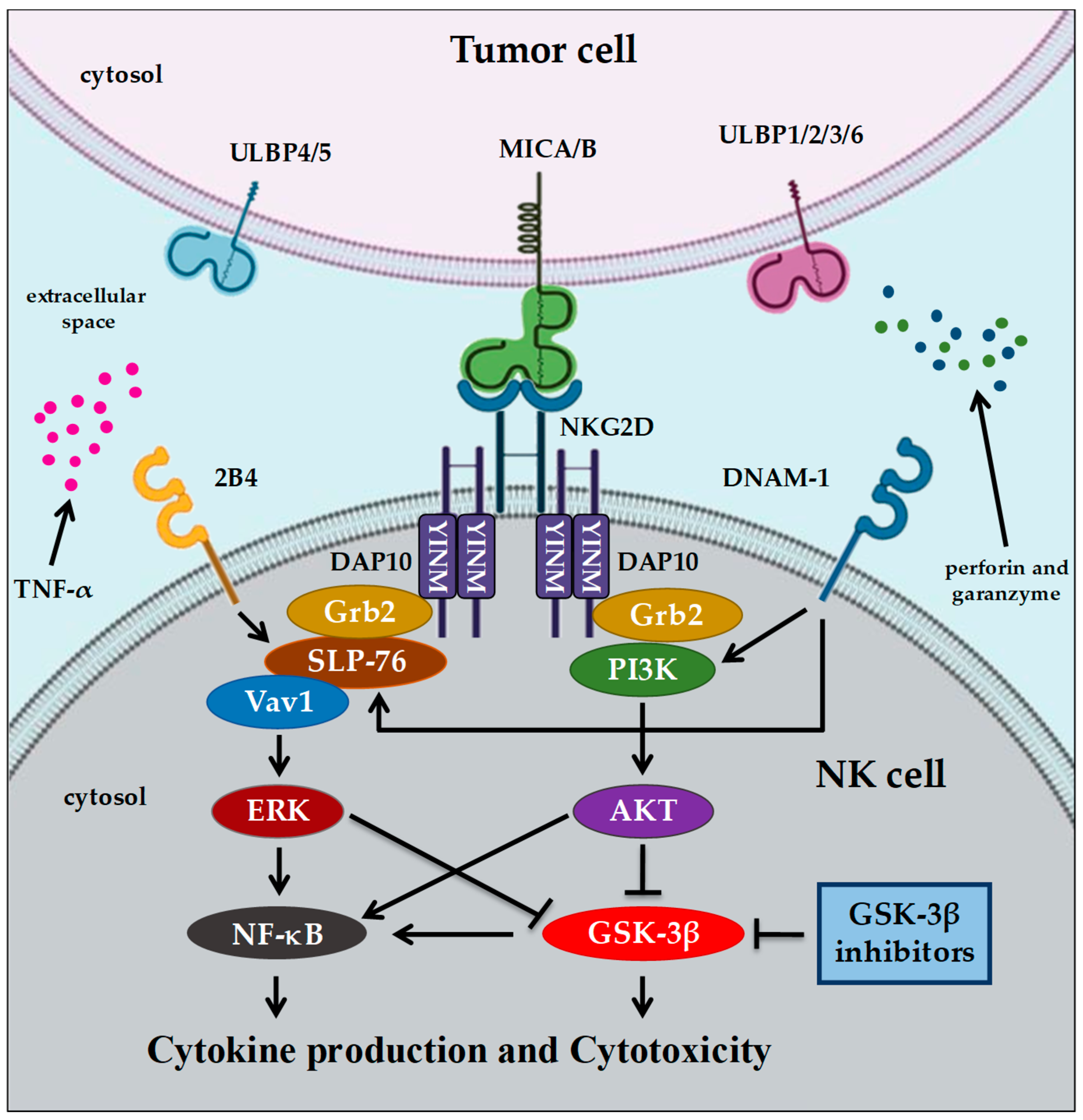
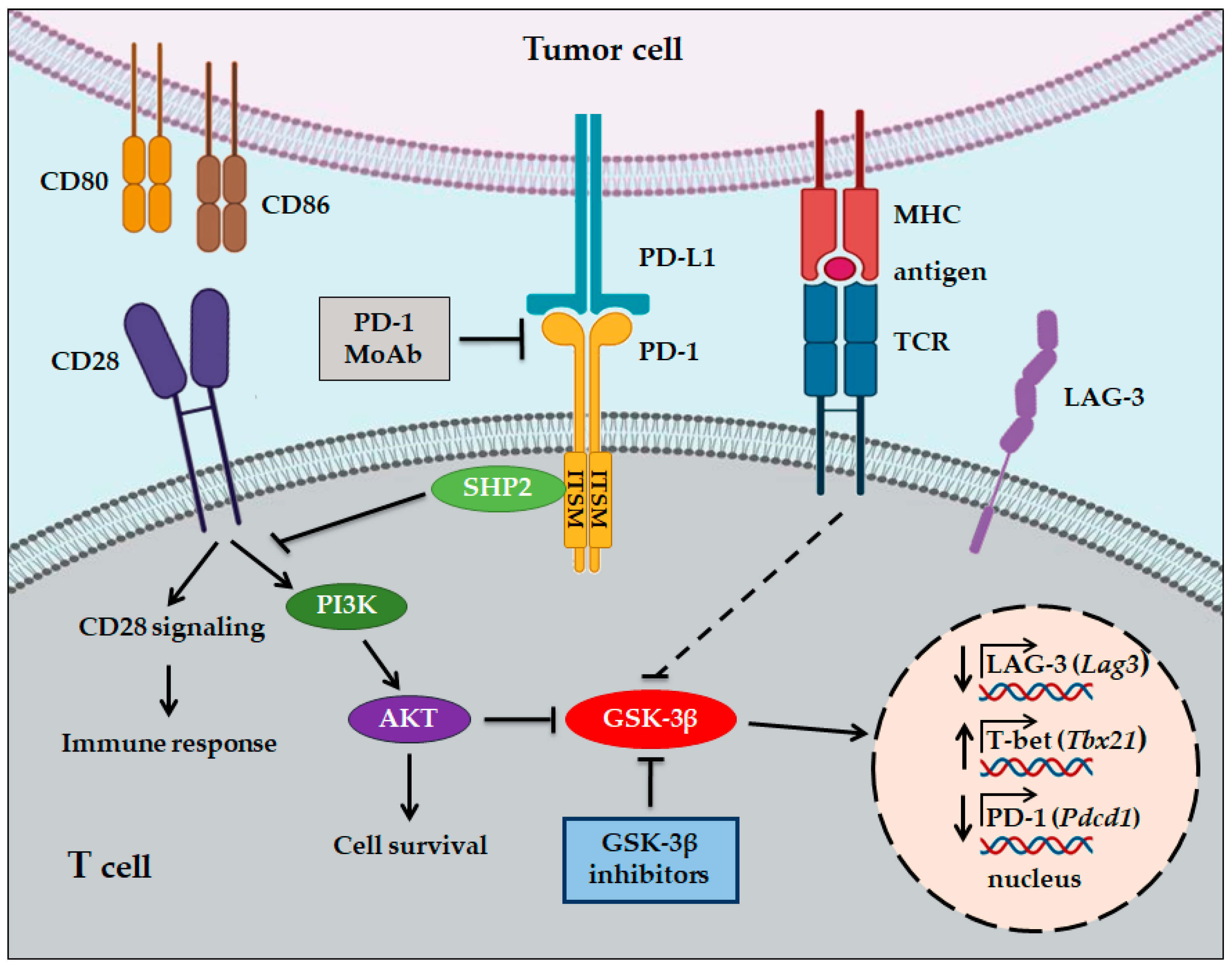
4. GSK-3 and Immunotherapy in Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cho, J.; Rameshwar, P.; Sadoshima, J. Distinct roles of glycogen synthase kinase (GSK)-3alpha and GSK-3beta in mediating cardiomyocyte differentiation in murine bone marrow-derived mesenchymal stem cells. J. Biol. Chem. 2009, 284, 36647–36658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3—At the crossroads of cell death and survival. J. Cell Sci. 2014, 127, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Carter, L.; Johnson, K.; Henry, R.R. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes 2002, 51, 2190–2198. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wolgamott, L.; Tcherkezian, J.; Vallabhapurapu, S.; Yu, Y.; Roux, P.P.; Yoon, S.O. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 2014, 33, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- De Matteis, S.; Napolitano, R.; Carloni, S. GSK-3β and its Unexpected Role in Immunity, Inflammation and Cancer. SM J. Oncol. Hematol. 2016, 1, 1001. [Google Scholar]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, C.; Bienz, M. Inhibition of GSK3 by Wnt signalling-two contrasting models. J. Cell Sci. 2011, 124, 3537–3544. [Google Scholar] [CrossRef] [Green Version]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Götschel, F.; Kern, C.; Lang, S.; Sparna, T.; Markmann, C.; Schwager, J.; McNelly, S.; von Weizsäcker, F.; Laufer, S.; Hecht, A.; et al. Inhibition of GSK3 differentially modulates NF-kappaB, CREB, AP-1 and beta-catenin signaling in hepatocytes, but fails to promote TNF-alpha-induced apoptosis. Exp. Cell Res. 2008, 14, 1351–1366. [Google Scholar] [CrossRef]
- Hua, F.; Zhou, J.; Liu, J.; Zhu, C.; Cui, B.; Lin, H.; Liu, Y.; Jin Yang, H.; Hu, Z. Glycogen synthase kinase-3beta negatively regulates TGF-beta1 and Angiotensin II-mediated cellular activity through interaction with Smad3. Eur. J. Pharmacol. 2010, 644, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, J.H.; Kim, Y.K.; Myoung, H.; Yun, P.Y. The role of tamoxifen in combination with cisplatin on oral squamous cell carcinoma cell lines. Cancer Lett. 2007, 245, 284–292. [Google Scholar] [CrossRef]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Lesort, M.; Jope, R.S.; Johnsn, G.V. Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 1999, 72, 576–584. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Shiraishi, F.; Hirata, M.; Miwa, Y.; Morimoto, S.; Sasaguri, T. Glycogen synthase kinase-3beta is tyrosine-phosphorylated by MEK1 in human skin fibroblasts. Biochem. Biophys. Res. Commun. 2004, 316, 411–415. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.W.; Clipstone, N.A. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J. Biol. Chem. 2001, 276, 3666–3673. [Google Scholar] [CrossRef] [Green Version]
- Grimes, C.A.; Jope, R.S. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. 2001, 78, 1219–1232. [Google Scholar] [CrossRef] [Green Version]
- Nikolakaki, E.; Coffer, P.J.; Hemelsoet, R.; Woodgett, J.R.; Defize, L.H. Glycogen synthase kinase 3 phosphorylates Jun family members in vitro and negatively regulates their transactivating potential in intact cells. Oncogene 1993, 8, 833–840. [Google Scholar]
- Medunjanin, S.; Schleithoff, L.; Fiegehenn, C.; Weinert, S.; Zuschratter, W.; Braun-Dullaeus, R.C. GSK-3β controls NF-kappaB activity via IKKγ/NEMO. Sci. Rep. 2016, 6, 38553. [Google Scholar] [CrossRef] [Green Version]
- Kulikov, R.; Boehme, K.A.; Blattner, C. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol. Cell. Biol. 2005, 25, 7170–7180. [Google Scholar] [CrossRef] [Green Version]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Purow, B. Notch inhibition as a promising new approach to cancer therapy. Adv. Exp. Med. Biol. 2012, 727, 305–319. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Foltz, D.R.; Santiago, M.C.; Berechid, B.E.; Nye, J.S. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr. Biol. 2002, 12, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Cervello, M.; Augello, G.; Cusimano, A.; Emma, M.R.; Balasus, D.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Pivotal roles of glycogen synthase-3 in hepatocellular carcinoma. Adv. Biol. Regul. 2017, 65, 59–76. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Richard, S.J.; Myoung-Sun, R. Glycogen Synthase Kinase-3 (GSK3) in Psychiatric Diseases and Therapeutic Interventions. Curr. Drug Targets 2006, 7, 1421–1434. [Google Scholar] [CrossRef]
- Sarhan, M.A.; Abdel-Hakeem, M.S.; Mason, A.L.; Tyrrell, D.L.; Houghton, M. Glycogen synthase kinase 3β inhibitors prevent hepatitis C virus release/assembly through perturbation of lipid metabolism. Sci. Rep. 2017, 7, 2495. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in Neurodegenerative Diseases. Int. J. Alzheimers Dis. 2011, 189246. [Google Scholar] [CrossRef] [Green Version]
- Mathuram, T.L.; Ravikumar, V.; Reece, L.M.; Karthik, S.; Sasikumar, C.S.; Cherian, K.M. Tideglusib induces apoptosis in human neuroblastoma IMR32 cells, provoking sub-G0/G1 accumulation and ROS generation. Environ. Toxicol. Pharmacol. 2016, 46, 194–205. [Google Scholar] [CrossRef]
- Ugolkov, A.; Qiang, W.; Bondarenko, G.; Procissi, D.; Gaisina, I.; James, C.D.; Chandler, J.; Kozikowski, A.; Gunosewoyo, H.; O’Halloran, T.; et al. Combination treatment with the GSK-3 inhibitor 9-ING-41 and CCNU cures orthotopic chemoresistant glioblastoma in patient-derived xenograft models. Transl. Oncol. 2017, 10, 669–678. [Google Scholar] [CrossRef]
- Mai, W.; Kawakami, K.; Shakoori, A.; Kyo, S.; Miyashita, K.; Yokoi, K.; Jin, M.; Shimasaki, T.; Motoo, Y.; Minamoto, T. Deregulated GSK3β sustains gastrointestinal cancer cells survival by modulating human telomerase reverse transcriptase and telomerase. Clin. Cancer Res. 2009, 15, 6810–6819. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Yamamoto, N.; Domoto, T.; Bolidong, D.; Hayashi, K.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Inatani, H.; Aoki, Y.; et al. Glycogen synthase kinase 3β as a potential therapeutic target in synovial sarcoma and fibrosarcoma. Cancer Sci. 2020, 111, 429–440. [Google Scholar] [CrossRef]
- Zeng, F.Y.; Dong, H.; Cui, J.; Liu, L.; Chen, T. Glycogen synthase kinase 3 regulates PAX3-FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2010, 391, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 37. [Google Scholar] [CrossRef] [Green Version]
- Kunnimalaiyaan, S.; Schwartz, V.K.; Jackson, I.A.; Clark Gamblin, T.; Kunnimalaiyaan, M. Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 2018, 18, 560. [Google Scholar] [CrossRef] [Green Version]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 Expression Inhibits YAP and TAZ Oncogenic Functions in Pancreatic Cancer. Mol. Cancer Ther. 2020, 19, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small-molecule glycogen synthase kinase-3 inhibitor, is active in neuroblastoma. Anticancer Drugs 2018, 29, 717–724. [Google Scholar] [CrossRef]
- Ding, L.; Madamsetty, V.S.; Kiers, S.; Alekhina, O.; Ugolkov, A.; Dube, J.; Zhang, Y.; Zhang, J.S.; Wang, E.; Dutta, S.K.; et al. Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to Chemotherapy by Abrogating the TopBP1/ATR-Mediated DNA Damage Response. Clin. Cancer Res. 2019, 25, 6452–6462. [Google Scholar] [CrossRef]
- Kuroki, H.; Anraku, T.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.P.; Giles, F.J.; Ugolkov, A.; Tomita, Y. 9-ING-41, a small molecule inhibitor of GSK-3beta, potentiates the effects of anticancer therapeutics in bladder cancer. Sci. Rep. 2019, 9, 19977. [Google Scholar] [CrossRef]
- Karmali, R.; Chukkapalli, V.; Gordon, L.I.; Borgia, J.A.; Ugolkov, A.; Mazar, A.P.; Giles, F.J. GSK-3β inhibitor, 9-ING-41, reduces cell viability and halts proliferation of B-cell lymphoma cell lines as a single agent and in combination with novel agents. Oncotarget 2017, 8, 114924–114934. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Stenson, M.; Abeykoon, J.; Nowakowski, K.; Zhang, L.; Lawson, J.; Wellik, L.; Li, Y.; Krull, J.; Wenzl, K.; et al. Targeting glycogen synthase kinase 3 for therapeutic benefit in lymphoma. Blood 2019, 134, 363–373. [Google Scholar] [CrossRef]
- Gray, J.E.; Infante, J.R.; Brail, L.H.; Simon, G.R.; Cooksey, J.F.; Jones, S.F.; Farrington, D.L.; Yeo, A.; Jackson, K.A.; Chow, K.H.; et al. A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Investig. New Drugs 2015, 33, 1187–1196. [Google Scholar] [CrossRef]
- Rizzieri, D.A.; Cooley, S.; Odenike, O.; Moonan, L.; Chow, K.H.; Jackson, K.; Wang, X.; Brail, L.; Borthakur, G. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk Lymphoma 2016, 57, 1800–1806. [Google Scholar] [CrossRef]
- Beldi-Ferchiou, A.; Caillat-Zucman, S. Control of NK Cell Activation by Immune Checkpoint Molecules. Int. J. Mol. Sci. 2017, 18, 2129. [Google Scholar] [CrossRef]
- Nirschl, C.J.; Drake, C.G. Molecular Pathways: Coexpression of Immune Checkpoint Molecules: Signaling Pathways and Implications for Cancer Immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [Green Version]
- Shifrin, N.; Raulet, D.H.; Ardolino, M. NK cell self tolerance, responsiveness and missing self recognition. Semin. Immunol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmberg, K.J.; Sohlberg, E.; Goodridge, J.P.; Ljunggren, H.G. Immune selection during tumor checkpoint inhibition therapy paves way for NK-cell “missing self” recognition. Immunogenetics 2017, 69, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses, integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Kim, H.S. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front. Immunol 2018, 9, 2041. [Google Scholar] [CrossRef]
- Boudreau, J.E.; Hsu, K.C. Natural killer cell education and the response to infection and cancer therapy: Stay tuned. Trends Immunol. 2018, 39, 222–239. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I–related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Shi, Y.; Vaziri, H.; Sharma, A.; Dhala, R.; Mills, G.B.; Miller, R.G. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J. Immunol. 1996, 156, 1788–1798. [Google Scholar]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-mediated signaling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef]
- Zheng, Y.; Collins, S.L.; Lutz, M.A.; Allen, A.N.; Kole, T.P.; Zarek, P.E.; Powell, J.D. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J. Immunol. 2007, 178, 2163–2170. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Kong, Q.; Kemp, K.; Zhao, Y.S.; Fang, D. Analysis of sirtuin 1 expression reveals a molecular explanation of IL-2-mediated reversal of T-cell tolerance. Proc. Natl. Acad. Sci. USA 2012, 109, 899–904. [Google Scholar] [CrossRef] [Green Version]
- Iida, T.; Ohno, H.; Nakaseko, C.; Sakuma, M.; Takeda-Ezaki, M.; Arase, H.; Kominami, E.; Fujisawa, T.; Saito, T. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J. Immunol. 2000, 165, 5062–5068. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Valk, E.; Leung, R.; Rudd, C.E. CTLA-4 Activation of Phosphatidylinositol 3-Kinase (PI 3-K) and Protein Kinase B (PKB/AKT) Sustains T-Cell Anergy without Cell Death. PLoS ONE 2008, 3, e3842. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef]
- Finger, L.R.; Pu, J.; Wasserman, R.; Vibhakar, R.; Louie, E.; Hardy, R.R.; Burrows, P.D.; Billips, L.G. The human PD-1 gene: Complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene 1997, 197, 177–187. [Google Scholar] [CrossRef]
- Chamoto, K.; Al-Habsi, M.; Honjo, T. Role of PD-1 in Immunity and Diseases. Curr. Top. Microbiol. Immunol. 2017, 410, 75–97. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Punt, C.J.; Hato, S.V.; Eleveld-Trancikova, D.; Jansen, B.J.; Nierkens, S.; Schreibelt, G.; de Boer, A.; Van Herpen, C.M.; Kaanders, J.H.; et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Investig. 2011, 121, 3100–3108. [Google Scholar] [CrossRef]
- Nazareth, M.R.; Broderick, L.; Simpson-Abelson, M.R.; Kelleher, R.J.; Yokota, S.J.; Bankert, R.B. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J. Immunol. 2007, 178, 5552–5562. [Google Scholar] [CrossRef] [Green Version]
- Messal, N.; Serriari, N.E.; Pastor, S.; Nunès, J.A.; Olive, D. PD-L2 is expressed on activated human T cells and regulates their function. Mol. Immunol. 2011, 48, 2214–2219. [Google Scholar] [CrossRef] [Green Version]
- Rozali, E.N.; Hato, S.V.; Robinson, B.W.; Lake, R.A.; Lesterhuis, W.J. Programmed Death Ligand 2 in Cancer-Induced Immune Suppression. Clin. Dev. Immunol. 2012, 656340. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Sahin, I.; Eturi, A.; De Souza, A.; Pamarthy, S.; Tavora, F.; Giles, F.J.; Carneiro, B.A. Glycogen synthase kinase-3 beta inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol. Ther. 2019, 20, 1047–1056. [Google Scholar] [CrossRef]
- Krueger, J.; Rudd, C.E.; Taylor, A. Glycogen synthase 3 (GSK-3) regulation of PD-1 expression and its therapeutic implications. Semin. Immunol. 2019, 42, 101295. [Google Scholar] [CrossRef]
- Fionda, C.; Malgarini, G.; Soriani, A.; Zingoni, A.; Cecere, F.; Iannitto, M.L.; Ricciardi, M.R.; Federico, V.; Petrucci, M.T.; Santoni, A.; et al. Inhibition of glycogen synthase kinase-3 increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of STAT3. J. Immunol. 2013, 190, 6662–6672. [Google Scholar] [CrossRef]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; deLima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7, 11154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.M.; White, M.J.; Goodier, M.R.; Riley, E.M. Functional Significance of CD57 Expression on Human NK Cells and Relevance to Disease. Front. Immunol. 2013, 4, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, B.; Cooley, S.; Verneris, M.R.; Curtsinger, J.; Luo, X.; Waller, E.K.; Anasetti, C.; Weisdorf, D.; Miller, J.S. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol. 2012, 189, 5082–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Vergès, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012, 119, 2665–2674. [Google Scholar] [CrossRef]
- Lopez-Vergès, S.; Milush, J.M.; Schwartz, B.S.; Pando, M.J.; Jarjoura, J.; York, V.A.; Houchins, J.P.; Miller, S.; Kang, S.M.; Norris, P.J.; et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 14725–14732. [Google Scholar] [CrossRef] [Green Version]
- Turkseven, M.R.; Oygur, T. Evaluation of natural killer cell defense in oral squamous cell carcinoma. Oral Oncol. 2010, 46, E34–E37. [Google Scholar] [CrossRef]
- Lv, L.; Pan, K.; Li, X.D.; She, K.L.; Zhao, J.J.; Wang, W.; Chen, J.G.; Chen, Y.B.; Yun, J.P.; Xia, J.C. The accumulation and prognosis value of tumor infiltrating IL-17 producing cells in esophageal squamous cell carcinoma. PLoS ONE 2011, 6, e18219. [Google Scholar] [CrossRef]
- Takanami, I.; Takeuchi, K.; Giga, M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J. Thorac. Cardiovasc. Surg. 2001, 121, 1058–1063. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 Inhibition Drives Maturation of NK Cells and Enhances Their Antitumor Activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [Green Version]
- Ohteki, T.; Parsons, M.; Zakarian, A.; Jones, R.G.; Nguyen, L.T.; Woodgett, J.R.; Ohashi, P. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J. Exp. Med. 2000, 192, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.; Schneider, H.; Rudd, C.E. cR and TcR-CD28 engagement of protein kinase B (PKB/AKT) and glycogen synthase kinase-3 (GSK-3) operates independently of guanine nucleotide exchange factor VAV-1. J. Biol. Chem. 2006, 281, 32385–32394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleman, L.J.; van Puijenbroek, A.A.; Shu, K.M.; Nadler, L.M.; Boussiotis, V.A. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J. Immunol. 2002, 168, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.; Rudd, C.E. Glycogen Synthase Kinase 3 Inactivation Compensates for the Lack of CD28 in the Priming of CD8+ Cytotoxic T-Cells: Implications for anti-PD-1 Immunotherapy. Front. Immunol. 2017, 8, 1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Zhuo, H.; Lu, Y.; Deng, L.; Jiang, R.; Zhang, L.; Zhu, Q.; Pu, L.; Wang, X.; Lu, L. Glycogen synthase kinase 3beta inhibition promotes human iTreg differentiation and suppressive function. Immunol. Res. 2015, 62, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhao, Y.L.; Lv, Y.P.; Cheng, P.; Chen, W.; Duan, M.; Teng, Y.S.; Wang, T.T.; Peng, L.S.; Mao, F.Y.; et al. Modulation of CD8+ memory stem T cell activity and glycogen synthase kinase 3β inhibition enhances anti-tumoral immunity in gastric cancer. Oncoimmunology 2018, 7, e1412900. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Guha, P.; Katz, S.; Sampath, P. Inhibition of GSK3beta leads to increased survival, proliferation and memory phenotype generation of GBM-specific CAR T cells. J. Immunol. 2015, 194. [Google Scholar] [CrossRef] [Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Cervello, M.; Emma, M.R.; Augello, G.; Cusimano, A.; Giannitrapani, L.; Soresi, M.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Gulino, A.; et al. New landscapes and horizons in hepatocellular carcinoma therapy. Aging 2020, 12, 3053–3094. [Google Scholar] [CrossRef]
- Taylor, A.; Harker, J.A.; Chanthong, K.; Stevenson, P.G.; Zuniga, E.I.; Rudd, C.E. Glycogen Synthase Kinase 3 Inactivation Drives T-bet-Mediated Downregulation of Co-receptor PD-to Enhance CD8+ Cytolytic T Cell Responses. Immunity 2016, 44, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, C.E.; Chanthong, K.; Taylor, A. Small Molecule Inhibition of GSK-3 Specifically Inhibits the Transcription of Inhibitory Co-receptor LAG-3 for Enhanced Anti-tumor Immunity. Cell Rep. 2020, 30, 2075–2082. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Katz, S.C.; Sengupta, S.; Sampath, P. Glycogen synthase kinase 3 inhibition lowers PD-1 expression, promotes long-term survival and memory generation in antigen-specific CAR-T cells. Cancer Lett. 2018, 433, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Subbiah, I.M.; Matos, I.; Garralda, E.; Menta, A.K.; Ganeshan, D.M.; Subbiah, V. Hyperprogression and Immunotherapy: Fact, Fiction, or Alternative Fact? Trends Cancer 2020, 6, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor Name | Tumor Type | Ref. |
|---|---|---|
| Tideglusib | glioblastoma | [34,35] |
| AR-A014418 | gastric cancer | [36] |
| synovial sarcoma | ||
| fibrosarcoma | [37] | |
| TWS119 | alveolar rhabdomyosarcoma | [38] |
| breast cancer | [39] | |
| LY2090314 | neuroblastoma | [40] |
| pancreatic cancer | [41] | |
| 9-ING-41 | glioblastoma | [35] |
| neuroblastoma | [42] | |
| pancreatic cancer | [43] | |
| bladder cancer | [44] | |
| lymphoma | [45,46] |
| Inhibitor Name | Therapeutic Application | Clinical Trials | Status |
|---|---|---|---|
| LY2090314 | metastatic cancer | NCT01287520 | Completed |
| acute leukemia | NCT01214603 | Completed | |
| pancreatic cancer | NCT01632306 | Terminated | |
| 9-ING-4 | lymphoma/pancreatic cancer | NCT03678883 | Recruiting |
| CHIR99021 | NK cells expanded in culture with CHIR99021 and IL-15, and then infused into patients with acute myelogenous leukemia (AML) | NCT03081780 | Active, not recruiting |
| CHIR99021 | NK cells expanded in culture with CHIR99021 and IL-15, and then infused into patients with ovarian cancer | NCT03213964 | Recruiting |
| CHIR99021 | NK cells expanded in culture with CHIR99021 and IL-15, and then infused into patients with solid tumors | NCT03319459 | Active, not recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Augello, G.; Emma, M.R.; Cusimano, A.; Azzolina, A.; Montalto, G.; McCubrey, J.A.; Cervello, M. The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells 2020, 9, 1427. https://doi.org/10.3390/cells9061427
Augello G, Emma MR, Cusimano A, Azzolina A, Montalto G, McCubrey JA, Cervello M. The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells. 2020; 9(6):1427. https://doi.org/10.3390/cells9061427
Chicago/Turabian StyleAugello, Giuseppa, Maria R. Emma, Antonella Cusimano, Antonina Azzolina, Giuseppe Montalto, James A. McCubrey, and Melchiorre Cervello. 2020. "The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment" Cells 9, no. 6: 1427. https://doi.org/10.3390/cells9061427
APA StyleAugello, G., Emma, M. R., Cusimano, A., Azzolina, A., Montalto, G., McCubrey, J. A., & Cervello, M. (2020). The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells, 9(6), 1427. https://doi.org/10.3390/cells9061427