In-Depth Investigation of Low-Abundance Proteins in Matured and Filling Stages Seeds of Glycine max Employing a Combination of Protamine Sulfate Precipitation and TMT-Based Quantitative Proteomic Analysis

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Plant Materials
2.2. Confirmation of Proteins, Amino Acids, and Fatty Acids Content in Seed Filling Stage Sample
2.3. Protein Extraction and Digestion by Filter-Aided Sample Preparation
2.4. TMT Labeling, Desalting, and BPRP Peptide Fractionation Using Stage-Tip
2.5. Q-Exactive MS Analysis
2.6. TMT Data Analysis by MaxQuant and Perseus Software
3. Results
3.1. TMT-Based Quantitative Proteomic Analysis of PS-Fractionated Mature Soybean Seed Samples
3.2. Functional Classification of Identified Proteins by PS Fractionation Method
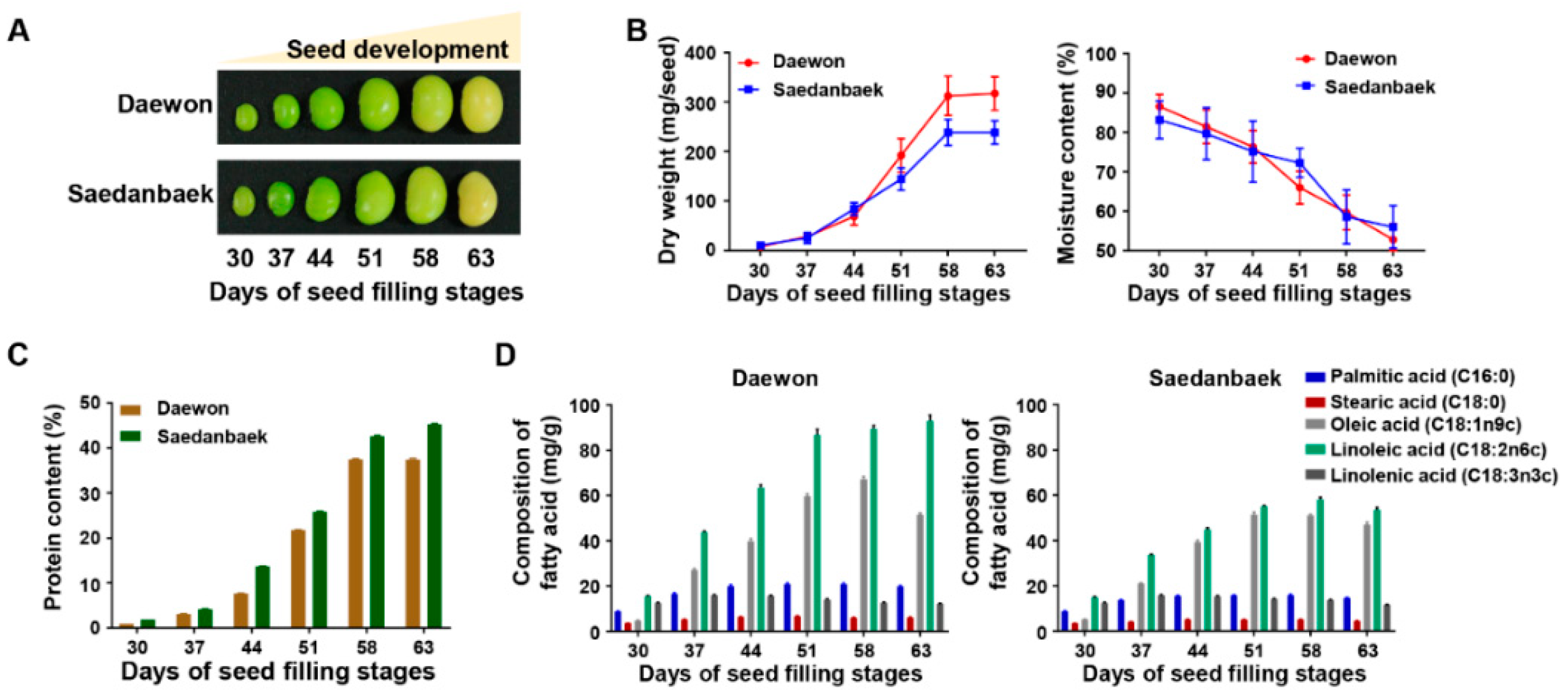
3.3. Physiological Validation and Free Amino Acids Analysis Using Seed Filling Stage Samples
3.4. Identifying the Proteome Changes between Two Varieties and Functional Annotation of Seed Filling Stage Samples
4. Discussion
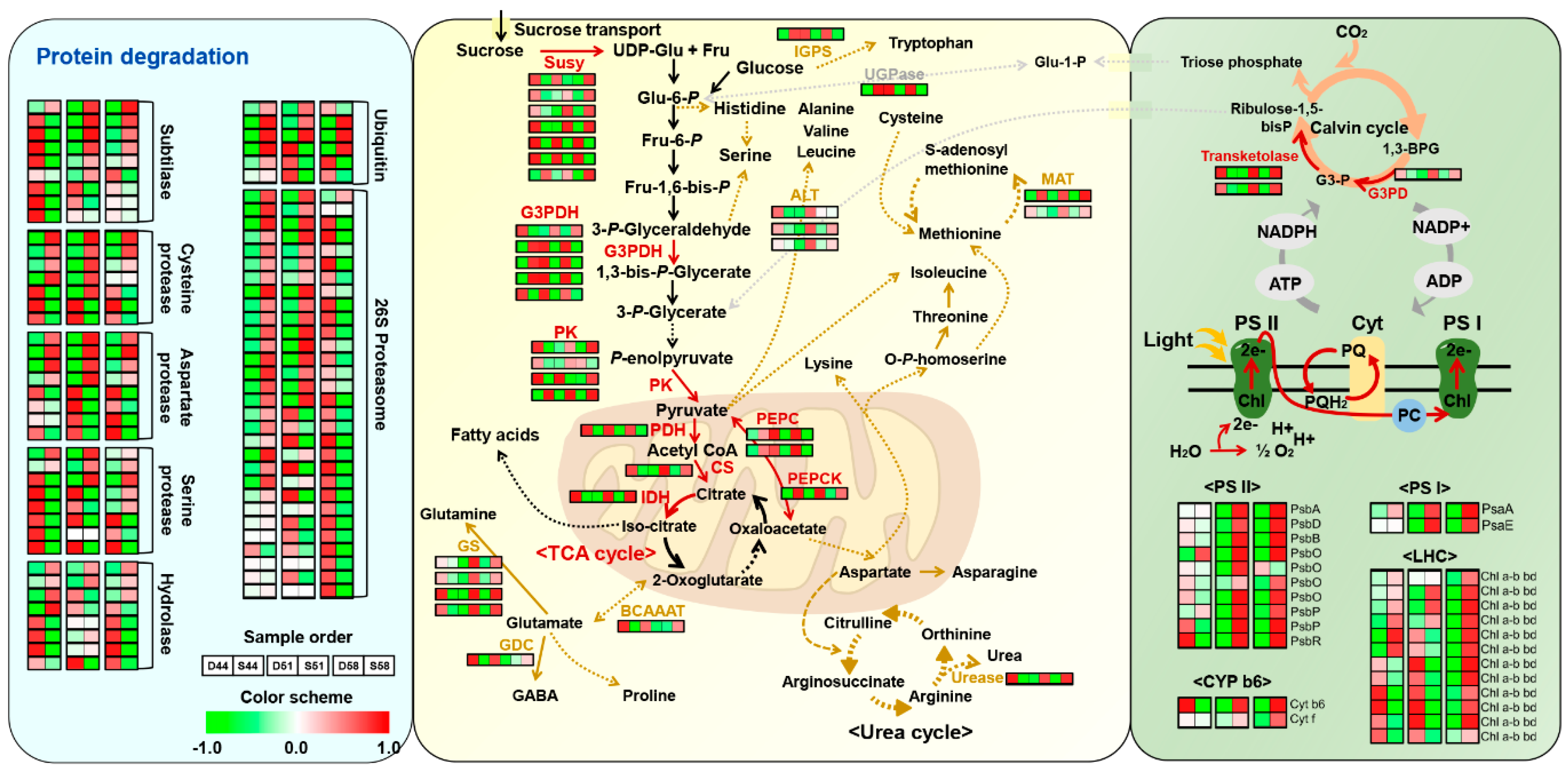
4.1. Enrichment of Proteins Participating in Major Metabolism in Seeds
4.2. Changes in the Free Amino Acids Profile Have a Positive Correlation with Protein Accumulation of Soybean Seeds during Seed Filling Stages
4.3. Differential Regulation of Photosynthesis and a Major Cho Metabolism between Two Varieties of Soybean Seeds during Filling Stages
4.4. Differential Regulation of Protein Degradation during Seed Filling Stages
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Note
References
- Tandang-Silvas, M.R.G.; Tecson-Mendoza, E.M.; Mikami, B.; Utsumi, S.; Maruyama, N. Molecular design of seed storage proteins for enhanced food physicochemical properties. Annu. Rev. Food Sci. Technol. 2011, 2, 59–73. [Google Scholar] [CrossRef]
- Min, C.W.; Gupta, R.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Concepts and strategies of soybean seed proteomics using the shotgun proteomics approach. Expert Rev. Proteomics 2019, 16, 795–804. [Google Scholar] [CrossRef]
- Wilson, K.A.; Rightmire, B.R.; Chen, J.C.; Tan-Wilson, A.L. Differential proteolysis of glycinin and β-conglycinin polypeptides during soybean germination and seedling growth. Plant Physiol. 1986, 82, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Lee, H.M.; Wang, Y.; Wu, J.; Kim, S.G.; Kang, K.Y.; Park, K.H.; Kim, Y.C.; Choi, I.S.; Agrawal, G.K.; et al. Depletion of abundant plant RuBisCO protein using the protamine sulfate precipitation method. Proteomics 2013, 13, 2176–2179. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wang, Y.; Gupta, R.; Kim, S.W.; Min, C.W.; Kim, Y.C.; Park, K.H.; Agrawal, G.K.; Rakwal, R.; Choung, M.G.; et al. Protamine sulfate precipitation method depletes abundant plant seed-storage proteins: A case study on legume plants. Proteomics 2015, 15, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.B.; Oehrle, N.W.; Natarajan, S.S. A rapid and simple procedure for the depletion of abundant storage proteins from legume seeds to advance proteome analysis: A case study using Glycine max. Proteomics 2009, 9, 3174–3188. [Google Scholar] [CrossRef]
- Mamontova, T.; Lukasheva, E.; Mavropolo-Solyarenko, G.; Proksch, C.; Bilova, T.; Kim, A.; Babakov, V.; Grishina, T.; Hoehenwarter, W.; Medvedev, S.; et al. Proteome map of Pea (Pisum sativum L.) embryos containing different amounts of residual chlorophylls. Int. J. Mol. Sci. 2018, 19, 4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [Green Version]
- Griffin, T.J.; Han, D.K.M.; Gygi, S.P.; Rist, B.; Lee, H.; Aebersold, R.; Parker, K.C. Toward a high-throughput approach to quantitative proteomic analysis: Expression-dependent protein identification by mass spectrometry. J. Am. Soc. Mass Spectrom. 2001, 12, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; He, Q.Y. Hunting Molecular Targets for Anticancer Reagents by Chemical Proteomics. In Novel Approaches and Strategies for Biologics, Vaccines and Cancer Therapies; Singh, M., Salnikova, M., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 347–363. [Google Scholar]
- Marx, H.; Minogue, C.E.; Jayaraman, D.; Richards, A.L.; Kwiecien, N.W.; Sihapirani, A.F.; Rajasekar, S.; Maeda, J.; Garcia, K.; Del Valle-Echevarria, A.R.; et al. A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. Nat. Biotechnol. 2016, 34, 1198–1205. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, S.; Wang, J.; Hu, Y. Quantitative proteomic analysis of wheat seeds during artificial ageing and priming using the isobaric tandem mass tag labeling. PLoS ONE 2016, 11, e0162851. [Google Scholar] [CrossRef]
- Min, C.W.; Kim, Y.J.; Gupta, R.; Kim, S.W.; Han, W.Y.; Ko, J.M.; Kang, H.W.; Yoon, W.B.; Choung, M.G.; Kim, Y.C.; et al. High-throughput proteome analysis reveals changes of primary metabolism and energy production under artificial aging treatment in Glycine max seeds. Appl. Biol. Chem. 2016, 59, 841–853. [Google Scholar] [CrossRef]
- Min, C.W.; Gupta, R.; Kim, S.W.; Lee, S.E.; Kim, Y.C.; Bae, D.W.; Han, W.Y.; Lee, B.W.; Ko, J.M.; Agrawal, G.K.; et al. Comparative biochemical and proteomic analyses of soybean seed cultivars differing in protein and oil content. J. Agric. Food Chem. 2015, 63, 7134–7142. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Xiao, H.; Zhang, Z.; Gao, X.; Zhao, J. Rapid and sensitive method for determining free amino acids in plant tissue by high-performance liquid chromatography with fluorescence detection. Acta Geochim. 2017, 36, 680–696. [Google Scholar] [CrossRef]
- Corleto, K.A.; Singh, J.; Jayaprakasha, G.K.; Patil, B.S. A sensitive HPLC-FLD method combined with multivariate analysis for the determination of amino acids in L-citrulline rich vegetables. J. Food Drug Anal. 2019, 27, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Min, C.W.; Kim, S.W.; Wang, Y.; Agrawal, G.K.; Rakwal, R.; Kim, S.G.; Lee, B.W.; Ko, J.M.; Baek, I.Y.; et al. Comparative investigation of seed coats of brown- versus yellow-colored soybean seeds using an integrated proteomics and metabolomics approach. Proteomics 2015, 15, 1706–1716. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Park, J.; Han, D.; Yang, J.; Kim, A.; Woo, J.; Kim, Y.; Mook-Jung, I. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 2018, 13, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Jin, J.; Woo, J.; Min, H.; Kim, Y. Proteomic analysis of mouse astrocytes and their secretome by a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics 2014, 14, 1604–1609. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Kramer, K.; Agrawal, G.K.; Rakwal, R.; Park, K.H.; Wang, Y.; Finkemeier, I.; Kim, S.T. A multi-omics analysis of Glycine max leaves reveals alteration in flavonoid and isoflavonoid metabolism upon ethylene and abscisic acid treatment. Proteomics 2018, 18, 1–10. [Google Scholar] [CrossRef]
- Pajarillo, E.A.B.; Kim, S.H.; Lee, J.Y.; Valeriano, V.D.V.; Kang, D.K. Quantitative proteogenomics and the reconstruction of the metabolic pathway in Lactobacillus mucosae LM1. Korean J. Food Sci. Anim. Resour. 2015, 35, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Min, C.W.; Lee, S.H.; Cheon, Y.E.; Han, W.Y.; Ko, J.M.; Kang, H.W.; Kim, Y.C.; Agrawal, G.K.; Rakwal, R.; Gupta, R.; et al. In-depth proteomic analysis of Glycine max seeds during controlled deterioration treatment reveals a shift in seed metabolism. J. Proteomics 2017, 169, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Plubell, D.L.; Wilmarth, P.A.; Zhao, Y.; Fenton, A.M.; Minnier, J.; Reddy, A.P.; Klimek, J.; Yang, X.; David, L.L.; Pamir, N. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell. Proteomics 2017, 16, 873–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Min, C.W.; Kim, Y.J.; Kim, S.T. Identification of Msp1-induced signaling components in rice leaves by integrated proteomic and phosphoproteomic analysis. Int. J. Mol. Sci. 2019, 20, 4135. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Min, C.W.; Kim, S.W.; Yoo, J.S.; Moon, A.-R.; Shin, A.-Y.; Kwon, S.-Y.; Kim, S.T. A TMT-based quantitative proteome analysis to elucidate the TSWV induced signaling cascade in susceptible and resistant cultivars of Solanum lycopersicum. Plants 2020, 9, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, H.B.; Coe, E.H. Seed Storage Proteins. In Encyclopedia of Genetics; Brenner, S., Miller, J.H., Eds.; Academic Press: New York, NY, USA, 2001; pp. 1782–1787. [Google Scholar]
- Natarajan, S.S.; Krishnan, H.B.; Lakshman, S.; Garrett, W.M. An efficient extraction method to enhance analysis of low abundant proteins from soybean seed. Anal. Biochem. 2009, 394, 259–268. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Wang, Y.; Kim, Y.C.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Expect the unexpected enrichment of “hidden proteome” of seeds and tubers by depletion of storage proteins. Front. Plant Sci. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, A.; Gaupels, F.; Sarioglu, H.; Holzmeister, C.; Spannagl, M.; Durner, J.; Lindermayr, C. Looking deep Inside: Detection of low-abundance proteins in leaf extracts of Arabidopsis and phloem exudates of pumpkin. Plant Physiol. 2012, 159, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.L.; Xiong, E.H.; An, S.F.; Gong, F.P.; Wang, W. Sequential extraction results in improved proteome profiling of medicinal plant pinellia ternata tubers, which contain large amounts of high-abundance proteins. PLoS ONE 2012, 7, e50497. [Google Scholar] [CrossRef]
- Xiong, E.; Wu, X.; Yang, L.; Gong, F.; Tai, F.; Wang, W. Chloroform-assisted phenol extraction improving proteome profiling of maize embryos through selective depletion of high-abundance storage proteins. PLoS ONE 2014, 9, e112724. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Gupta, R.; Kim, S.H.; Wang, Y.; Rakwal, R.; Agrawal, G.K.; Kim, S.T. Abundant storage protein depletion from tuber proteins using ethanol precipitation method: Suitability to proteomics study. Proteomics 2015, 15, 1765–1769. [Google Scholar] [CrossRef]
- Kim, S.T.; Cho, K.S.; Jang, Y.S.; Kang, K.Y. Two-dimensional electrophoretic analysis of rice proteins by polyethylene glycol fractionation for protein arrays. Electrophoresis 2001, 22, 2103–2109. [Google Scholar] [CrossRef]
- Miernyk, J.A.; Johnston, M.L. Chemical Cross-Linking Immobilized Concanavalin A for use in Proteomic Analyses. Prep. Biochem. Biotechnol. 2006, 36, 203–214. [Google Scholar] [CrossRef]
- Righetti, P.G.; Boschetti, E. Global proteome analysis in plants by means of peptide libraries and applications. J. Proteomics 2016, 143, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Q.; Song, B.-Y.; Deng, Z.-J.; Wang, Y.; Liu, S.-J.; Møller, I.M.; Song, S.-Q. Proteomic analysis of lettuce seed germination and thermoinhibition by sampling of individual seeds at germination and removal of storage proteins by polyethylene glycol fractionation. Plant Physiol. 2015, 167, 1332–1350. [Google Scholar] [CrossRef] [PubMed]
- Vernoud, V.; Horton, A.C.; Yang, Z.; Nielsen, E. Analysis of the small GTPase gene superfamily of arabidopsis. Plant Physiol. 2003, 131, 1191–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, L.; Abildgaard, A.B.; Gersing, S.K.; Stein, A.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Protein stability and degradation in health and disease. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: San Diego, CA, USA, 2019; Volume 114, pp. 61–83. [Google Scholar]
- Gallardo, K.; Thompson, R.; Burstin, J. Reserve accumulation in legume seeds. Comptes Rendus Biol. 2008, 331, 755–762. [Google Scholar] [CrossRef]
- Weber, H.; Borisjuk, L.; Wobus, U. Molecular physiology of legume seed development. Annu. Rev. Plant Biol. 2005, 56, 253–279. [Google Scholar] [CrossRef]
- Amir, R.; Galili, G.; Cohen, H. The metabolic roles of free amino acids during seed development. Plant Sci. 2018, 275, 11–18. [Google Scholar] [CrossRef]
- Ohyama, T.; Ohtake, N.; Sueyoshi, K.; Ono, Y.; Tsutsumi, K.; Ueno, M.; Tanabata, S.; Sato, T.; Takahashi, Y. Amino Acid Metabolism and Transport in Soybean Plants. In Amino Acid—New Insights and Roles in Plant and Animal; Toshiki, A., Md, A., Eds.; IntechOpen: London, UK, 2017; pp. 171–196. [Google Scholar]
- Pandurangan, S.; Pajak, A.; Molnar, S.J.; Cober, E.R.; Dhaubhadel, S.; Hernández-Sebasti, C.; Kaiser, W.M.; Nelson, R.L.; Huber, S.C.; Marsolais, F. Relationship between asparagine metabolism and protein concentration in soybean seed. J. Exp. Bot. 2012, 63, 3173–3184. [Google Scholar] [CrossRef]
- Frank, A.; Cohen, H.; Hoffman, D.; Amir, R. Methionine and S-methylmethionine exhibit temporal and spatial accumulation patterns during the Arabidopsis life cycle. Amino Acids 2015, 47, 497–510. [Google Scholar] [CrossRef]
- Tan, H.; Xie, Q.; Xiang, X.; Li, J.; Zheng, S.; Xu, X.; Guo, H.; Ye, W. Dynamic metabolic profiles and tissue-specific source effects on the metabolome of developing seeds of Brassica napus. PLoS ONE 2015, 10, e0124794. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Todd, C.D.; Trovato, M.; Forlani, G.; Funck, D. Physiological implications of arginine metabolism in plants. Front. Plant Sci. 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Micallef, B.J.; Shelp, B.J. Arginine metabolism in developing soybean cotyledons. Plant Physiol. 1989, 90, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Schneidereit, J.; Häusler, R.E.; Fiene, G.; Kaiser, W.M.; Weber, A.P.M. Antisense repression reveals a crucial role of the plastidic 2-oxoglutarate/malate translocator DiT1 at the interface between carbon and nitrogen metabolism. Plant J. 2006, 45, 206–224. [Google Scholar] [CrossRef]
- Amir, R. Current understanding of the factors regulating methionine content in vegetative tissues of higher plants. Amino Acids 2010, 39, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Pajak, A.; Pandurangan, S.; Amir, R.; Marsolais, F. Higher endogenous methionine in transgenic Arabidopsis seeds affects the composition of storage proteins and lipids. Amino Acids 2016, 48, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Hou, W.; Godo, I.; Wu, C.; Yu, Y.; Matityahu, I.; Hacham, Y.; Sun, S.; Han, T.; Amir, R. Soybean seeds expressing feedback-insensitive cystathionine γ-synthase exhibit a higher content of methionine. J. Exp. Bot. 2013, 64, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Galili, G.; Avin-Wittenberg, T.; Angelovici, R.; Fernie, A.R. The role of photosynthesis and amino acid metabolism in the energy status during seed development. Front. Plant Sci. 2014, 5, 1–6. [Google Scholar] [CrossRef]
- Borisjuk, L.; Nguyen, T.H.; Neuberger, T.; Rutten, T.; Tschiersch, H.; Claus, B.; Feussner, I.; Webb, A.G.; Jakob, P.; Weber, H.; et al. Gradients of lipid storage, photosynthesis and plastid differentiation in developing soybean seeds. New Phytol. 2005, 167, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Malone, S.; Chen, Z.H.; Bahrami, A.R.; Walker, R.P.; Gray, J.E.; Leegood, R.C. Phosphoenolpyruvate carboxykinase in arabidopsis: Changes in gene expression, protein and activity during vegetative and reproductive development. Plant Cell Physiol. 2007, 48, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Leegood, R.C.; Ap Rees, T. Phosphoenolpyruvate carboxykinase and gluconeogenesis in cotyledons of Cucurbita pepo. BBA Enzymol. 1978, 524, 207–218. [Google Scholar] [CrossRef]
- Walker, R.P.; Trevanion, S.J.; Leegood, R.C. Phosphoenolpyruvate carboxykinase from higher plants: Purification from cucumber and evidence of rapid proteolytic cleavage in extracts from a range of plant tissues. Planta 1995, 196, 58–63. [Google Scholar] [CrossRef]
- Delgado-Alvarado, A.; Walker, R.P.; Leegood, R.C. Phosphoenolpyruvate carboxykinase in developing pea seeds is associated with tissues involved in solute transport and is nitrogen-responsive. Plant, Cell Environ. 2007, 30, 225–235. [Google Scholar] [CrossRef]
- Gallardo, K.; Firnhaber, C.; Zuber, H.; Héricher, D.; Belghazi, M.; Henry, C.; Küster, H.; Thompson, R. A combined proteome and ranscriptome analysis of developing Medicago truncatula seeds: Evidence for metabolic specialization of maternal and filial tissues. Mol. Cell. Proteomics 2007, 6, 2165–2179. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, K.; Kurt, C.; Thompson, R.; Ochatt, S. In vitro culture of immature M. truncatula grains under conditions permitting embryo development comparable to that observed in vivo. Plant Sci. 2006, 170, 1052–1058. [Google Scholar] [CrossRef]
- Sharma, B.; Joshi, D.; Yadav, P.K.; Gupta, A.K.; Bhatt, T.K. Role of ubiquitin-mediated degradation system in plant biology. Front. Plant Sci. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Deruyffelaere, C.; Bouchez, I.; Morin, H.; Guillot, A.; Miquel, M.; Froissard, M.; Chardot, T.; D’Andrea, S. Ubiquitin-mediated proteasomal degradation of oleosins is involved in oil body mobilization during post-germinative seedling growth in Arabidopsis. Plant Cell Physiol. 2015, 56, 1374–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macho, A.P.; Zipfel, C. Plant PRRs and the activation of innate immune signaling. Mol. Cell 2014, 54, 263–272. [Google Scholar] [CrossRef]
- Santner, A.; Estelle, M. The ubiquitin-proteasome system regulates plant hormone signaling. Plant J. 2010, 61, 1029–1040. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, C.W.; Park, J.; Bae, J.W.; Agrawal, G.K.; Rakwal, R.; Kim, Y.; Yang, P.; Kim, S.T.; Gupta, R. In-Depth Investigation of Low-Abundance Proteins in Matured and Filling Stages Seeds of Glycine max Employing a Combination of Protamine Sulfate Precipitation and TMT-Based Quantitative Proteomic Analysis. Cells 2020, 9, 1517. https://doi.org/10.3390/cells9061517
Min CW, Park J, Bae JW, Agrawal GK, Rakwal R, Kim Y, Yang P, Kim ST, Gupta R. In-Depth Investigation of Low-Abundance Proteins in Matured and Filling Stages Seeds of Glycine max Employing a Combination of Protamine Sulfate Precipitation and TMT-Based Quantitative Proteomic Analysis. Cells. 2020; 9(6):1517. https://doi.org/10.3390/cells9061517
Chicago/Turabian StyleMin, Cheol Woo, Joonho Park, Jin Woo Bae, Ganesh Kumar Agrawal, Randeep Rakwal, Youngsoo Kim, Pingfang Yang, Sun Tae Kim, and Ravi Gupta. 2020. "In-Depth Investigation of Low-Abundance Proteins in Matured and Filling Stages Seeds of Glycine max Employing a Combination of Protamine Sulfate Precipitation and TMT-Based Quantitative Proteomic Analysis" Cells 9, no. 6: 1517. https://doi.org/10.3390/cells9061517
APA StyleMin, C. W., Park, J., Bae, J. W., Agrawal, G. K., Rakwal, R., Kim, Y., Yang, P., Kim, S. T., & Gupta, R. (2020). In-Depth Investigation of Low-Abundance Proteins in Matured and Filling Stages Seeds of Glycine max Employing a Combination of Protamine Sulfate Precipitation and TMT-Based Quantitative Proteomic Analysis. Cells, 9(6), 1517. https://doi.org/10.3390/cells9061517