Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection


Abstract
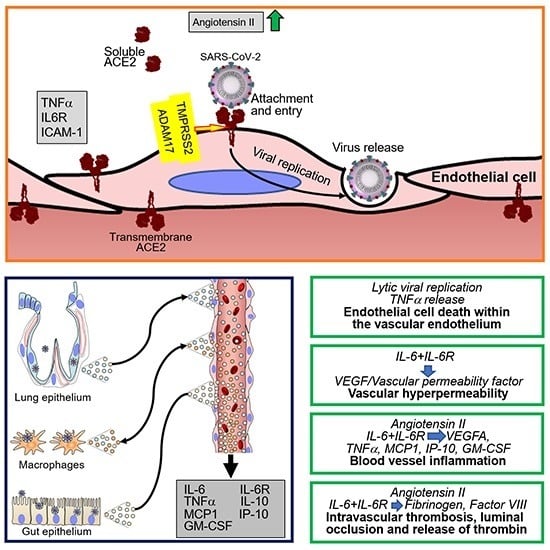
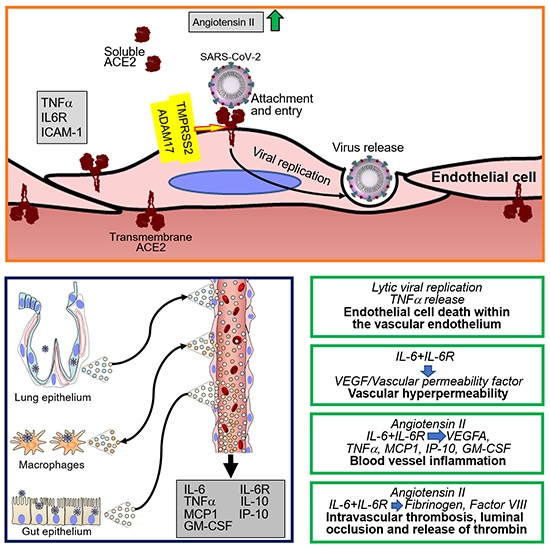
:
1. Introduction
2. Functions of the Angiotensin-Converting Enzyme 2 (ACE2)
2.1. ACE2, a Regulator of the Renin-Angiotensin System
2.2. Functions of ACE2 in Endothelial Cells
2.3. Non-Vascular Functions of ACE2
3. ACE2 Functions as a Coronavirus Receptor
3.1. ACE2 Is a Receptor for SARS-CoV-2 and other Coronaviruses
3.2. ACE2 Internalization and Shedding by Coronaviruses
4. SARS-CoV-2 Infection of Endothelial Cells and Vascular Pathology
4.1. SARS-CoV-2 Infection of Endothelial Cells
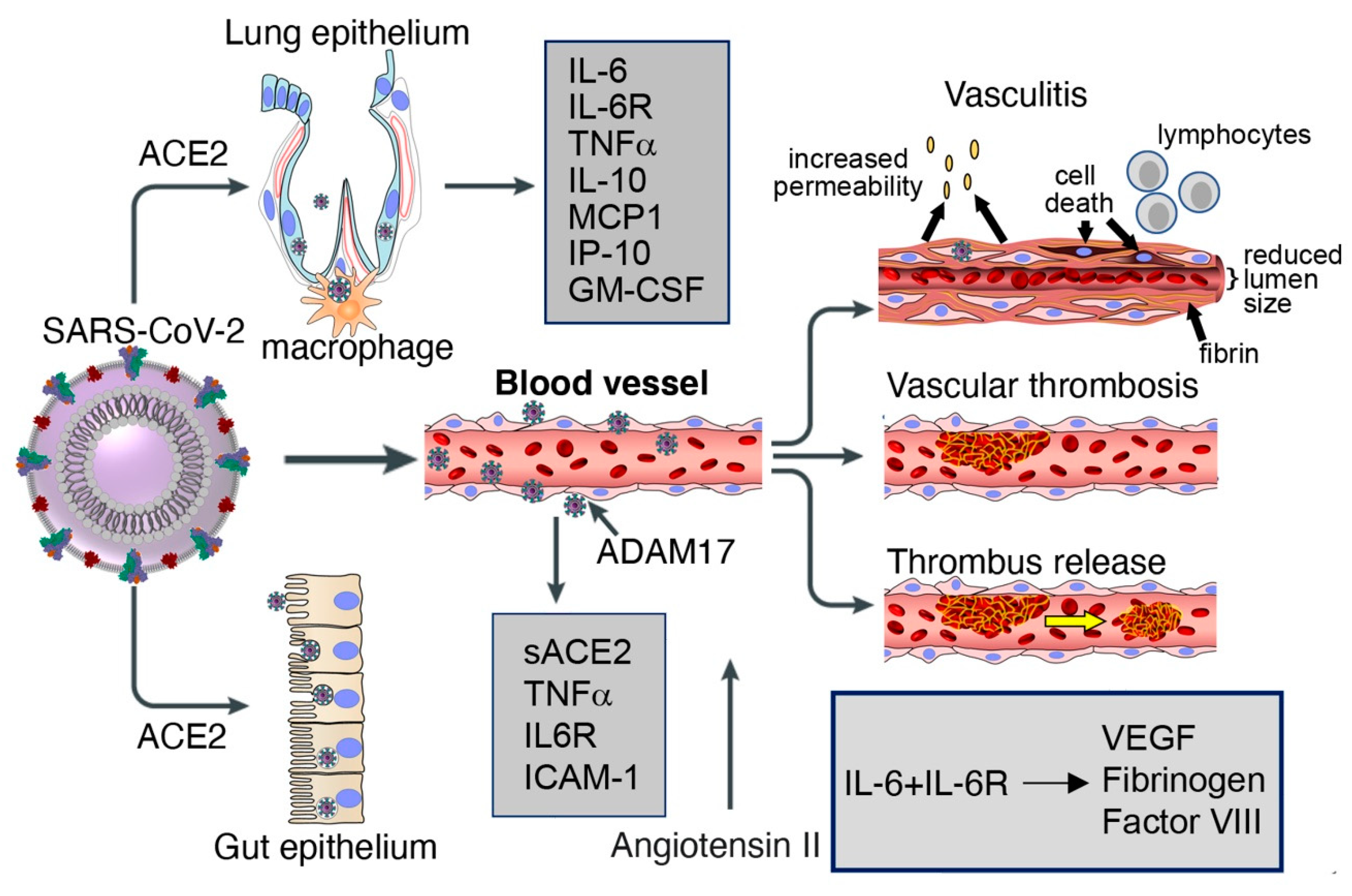
4.2. Vascular Pathology in Patients Infected with SARS-CoV-2
4.3. Cytokines and Coagulation Profile in COVID-19
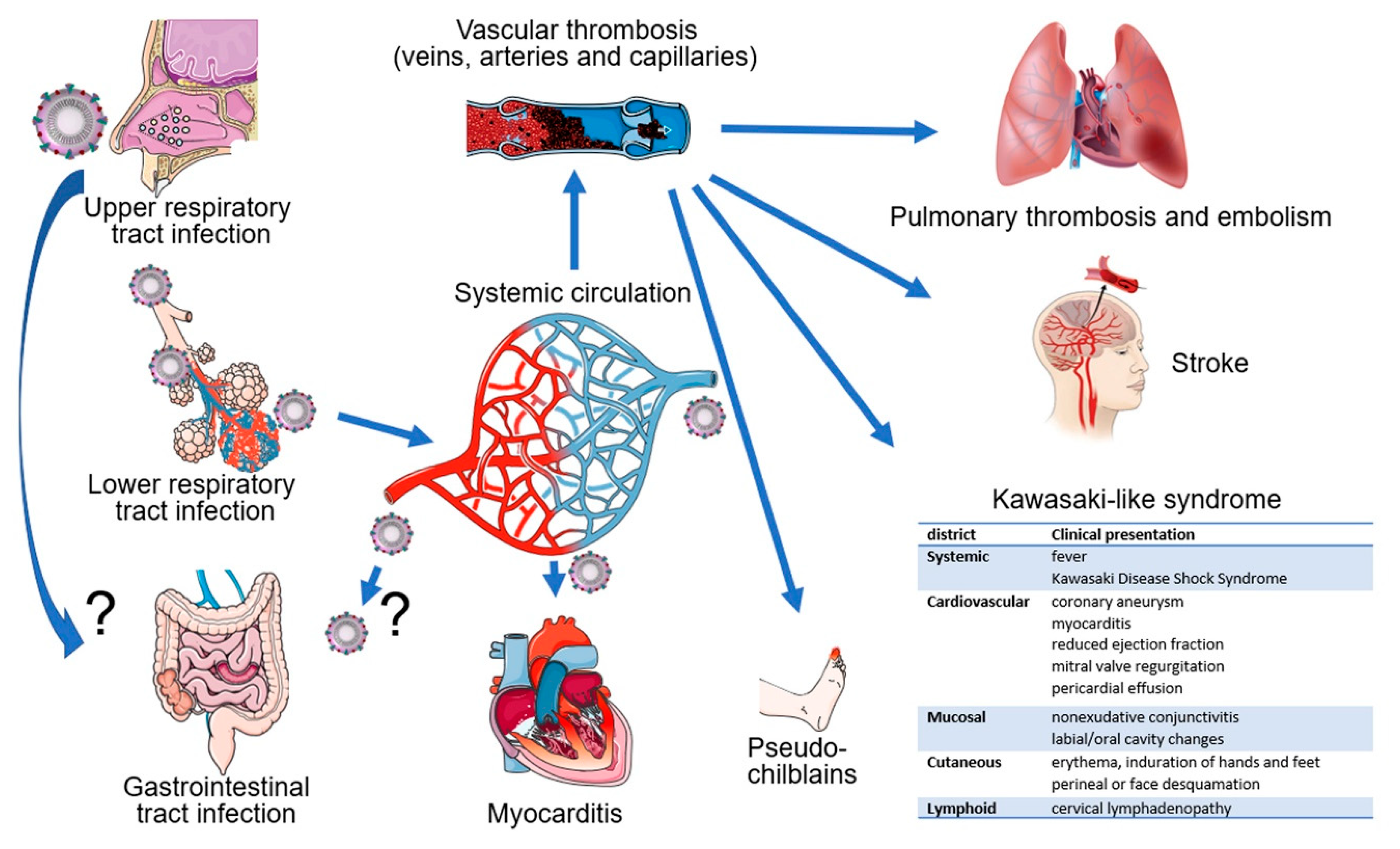
5. Vascular Manifestations of COVID-19
5.1. Demographics of COVID-19 Patients
5.2. Pulmonary Disease
5.3. Gastrointestinal Disease
5.4. Neurological Disease
5.5. Myocardial Disease
5.6. Cutaneous Disease
5.7. Kawasaki-Like Syndrome/Multisystem Inflammatory Syndrome in Children (MIS-C)
6. Insights into Factors that Precipitate Vascular Pathology in COVID-19
7. Approaches to the Prevention and Management of Vascular Manifestations in COVID-19
8. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Donnelly, C.A.; Malik, M.R.; Elkholy, A.; CauChemez, S.; Van Kerkhove, M.D. Worldwide Reduction in MERS Cases and Deaths since 2016. Emerg. Infect. Dis. 2019, 25, 1758–1760. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Micro Biol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Rajgor, D.D.; Lee, M.H.; Archuleta, S.; Bagdasarian, N.; Quek, S.C. The many estimates of the COVID-19 case fatality rate. Lancet Infect. Dis. 2020, 20, 776–777. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Shimizu, H.; Shibue, Y.; Hosoda, T.; Iwabuchi, K.; Nagamine, K.; Saito, H.; Sawada, R.; Oishi, T.; Tsukiji, J.; et al. Clinical course of 2019 novel coronavirus Disease (COVID-19) in individuals present during the outbreak on the Diamond Princess cruise ship. J. Infect. Chemother. 2020, 26, 865–869. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Boehm, M.; Nabel, E.G. Angiotensin-converting enzyme 2—A new cardiac regulator. N. Engl. J. Med. 2002, 347, 1795–1797. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Crackower, M.A.; Backx, P.H.; Penninger, J.M. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc. Med. 2003, 13, 93–101. [Google Scholar] [CrossRef]
- Huentelman, M.J.; Zubcevic, J.; Katovich, M.J.; Raizada, M.K. Cloning and characterization of a secreted form of angiotensin-converting enzyme 2. Regul. Pept. 2004, 122, 61–67. [Google Scholar] [CrossRef]
- Turner, A.J.; Tipnis, S.R.; Guy, J.L.; Rice, G.; Hooper, N.M. ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors. Can. J. Physiol. Pharmacol. 2002, 80, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.J. Exploring the structure and function of zinc metallopeptidases: Old enzymes and new discoveries. Biochem. Soc. Trans. 2003, 31, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Signaling by the angiotensin-converting enzyme. Circ. Res. 2006, 98, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Danilczyk, U.; Eriksson, U.; Crackower, M.A.; Penninger, J.M. A story of two ACEs. J. Mol. Med. 2003, 81, 227–234. [Google Scholar] [CrossRef]
- Clarke, N.E.; Turner, A.J. Angiotensin-converting enzyme 2: The first decade. Int. J. Hypertens 2012, 2012, 307315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Trask, A.J.; Jessup, J.A. Advances in bioChem.ical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2281–H2290. [Google Scholar] [CrossRef] [Green Version]
- Warner, F.J.; Lew, R.A.; Smith, A.I.; Lambert, D.W.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J. Biol. Chem. 2005, 280, 39353–39362. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Jones, A.L.; Grant, P.J.; Carter, A.M.; Turner, A.J.; Hooper, N.M. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006, 48, 914–920. [Google Scholar] [CrossRef]
- Lew, R.A.; Warner, F.J.; Hanchapola, I.; Yarski, M.A.; Ramchand, J.; Burrell, L.M.; Smith, A.I. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp. Physiol. 2008, 93, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Uri, K.; Fagyas, M.; Manyine Siket, I.; Kertesz, A.; Csanadi, Z.; Sandorfi, G.; Clemens, M.; Fedor, R.; Papp, Z.; Edes, I.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE 2014, 9, e87845. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.H. Soluble angiotensin-converting enzyme 2 in human heart failure: Relation with myocardial function and clinical outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soro-Paavonen, A.; Gordin, D.; Forsblom, C.; Rosengard-Barlund, M.; Waden, J.; Thorn, L.; Sandholm, N.; Thomas, M.C.; Groop, P.H.; FinnDiane Study, G. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J. Hypertens 2012, 30, 375–383. [Google Scholar] [CrossRef]
- Soler, M.J.; Ye, M.; Wysocki, J.; William, J.; Lloveras, J.; Batlle, D. Localization of ACE2 in the renal vasculature: Amplification by angiotensin II type 1 receptor blockade using telmisartan. Am. J. Physiol. Renal. Physiol. 2009, 296, F398–F405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano, L.; Riera, M.; Pascual, J.; Soler, M.J. Circulating ACE2 in Cardiovascular and Kidney Diseases. Curr. Med. Chem. 2017, 24, 3231–3241. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E. Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ. Res. 2017, 121, 43–55. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.A.; Velkoska, E.; Ierino, F.L.; Burrell, L.M. Angiotensin-converting enzyme 2 activity in patients with chronic kidney Disease. Nephrol. Dial. Transplant. 2013, 28, 2287–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhang, P.; Liang, T.; Chen, Y.; Liu, D.; Yu, H. Relationship between circulating levels of angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis and coronary heart Disease. Hear. Vessel. 2020, 35, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Sriramula, S.; Chhabra, K.H.; Lazartigues, E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ. Res. 2013, 113, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002, 532, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, H.B.; Lyu, J.R.; Lei, X.M.; Li, W.; Wu, G.; Lyu, J.; Dai, Z.M. Specific ACE2 Expression in Small Intestinal Enterocytes may Cause Gastrointestinal Symptoms and Injury after 2019-nCoV Infection. Int. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue Dis.tribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Tang, M.; Zheng, X.; Liu, Y.; Li, X.; Shan, H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 2020, 158, 1831–1833.e3. [Google Scholar] [CrossRef]
- Tikellis, C.; Johnston, C.I.; Forbes, J.M.; Burns, W.C.; Burrell, L.M.; Risvanis, J.; Cooper, M.E. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension 2003, 41, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, J.; Xu, Y.-P.; Zhou, T.-L.; Jin, Y.-Y.; Lou, J.-N. Expression of severe acute respiratory syndrome coronavirus receptors, ACE2 and CD209L in different organ derived microvascular endothelial cells. Zhonghua Yi Xue Za Zhi 2007, 87, 833–837. [Google Scholar]
- Weir, M.R.; Dzau, V.J. The renin-angiotensin-aldosterone system: A specific target for hypertension management. Am. J. Hypertens 1999, 12, 205S–213S. [Google Scholar] [CrossRef]
- Eguchi, S.; Kawai, T.; Scalia, R.; Rizzo, V. Understanding Angiotensin II Type 1 Receptor Signaling in Vascular Pathophysiology. Hypertension 2018, 71, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R.; Recinos, A., 3rd; Eledrisi, M.S. Vascular inflammation and the renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.; Ferreira, A.J. Pharmacological effects of AVE 0991, a nonpeptide angiotensin-(1-7) receptor agonist. Cardiovasc. Drug Rev. 2006, 24, 239–246. [Google Scholar] [CrossRef]
- Nickenig, G.; Harrison, D.G. The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part II: AT(1) receptor regulation. Circulation 2002, 105, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [Green Version]
- Pena Silva, R.A.; Chu, Y.; Miller, J.D.; Mitchell, I.J.; Penninger, J.M.; Faraci, F.M.; Heistad, D.D. Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke 2012, 43, 3358–3363. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Pickering, R.J.; Tsorotes, D.; Koitka, A.; Sheehy, K.; Bernardi, S.; Toffoli, B.; Nguyen-Huu, T.P.; Head, G.A.; Fu, Y.; et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 2010, 107, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, Y.; Zeng, Z.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-kappaB pathways. Sci. Rep. 2015, 5, 8209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, X.; Hou, Z.; Deng, F. RhACE2—playing an important role in inhibiting apoptosis induced by Ang II in HUVECs. Medicine (Baltimore) 2019, 98, e15799. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Carretero, O.A.; Xu, J.; Harding, P.; Ramadurai, N.; Gu, X.; Peterson, E.; Yang, X.P. Activation of angiotensin II type 2 receptor suppresses TNF-alpha-induced ICAM-1 via NF-small ka, CyrillicB: Possible role of ACE2. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H827–H834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, J.; Luo, J.Y.; Tian, X.Y.; Cheang, W.S.; Xu, J.; Lau, C.W.; Wang, L.; Wong, W.T.; Wong, C.M.; et al. Upregulation of Angiotensin (1-7)-Mediated Signaling Preserves Endothelial Function Through Reducing Oxidative Stress in Diabetes. Antioxid Redox Signal. 2015, 23, 880–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Gao, F.; Sun, B.; Hao, J.; Liu, Z. Angiotensin-Converting Enzyme 2 Inhibits Apoptosis of Pulmonary Endothelial Cells During Acute Lung Injury Through Suppressing SMAD2 Phosphorylation. Cell. Physiol. Biochem. 2015, 35, 2203–2212. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Gao, F.; Xie, G.; Liu, Z. Angiotensin-Converting Enzyme 2 Inhibits Apoptosis of Pulmonary Endothelial Cells During Acute Lung Injury Through Suppressing MiR-4262. Cell. Physiol. Biochem. 2015, 37, 759–767. [Google Scholar] [CrossRef]
- Yu, X.; Lin, Q.; Qin, X.; Ruan, Z.; Zhou, J.; Lin, Z.; Su, Y.; Zheng, J.; Liu, Z. ACE2 Antagonizes VEGFa to Reduce Vascular Permeability During Acute Lung Injury. Cell. Physiol. Biochem. 2016, 38, 1055–1062. [Google Scholar] [CrossRef]
- Kaminska, M.; Mogielnicki, A.; Stankiewicz, A.; Kramkowski, K.; Domaniewski, T.; Buczko, W.; Chabielska, E. Angiotensin II via AT1 receptor accelerates arterial thrombosis in renovascular hypertensive rats. J. Physiol. Pharmacol. 2005, 56, 571–585. [Google Scholar]
- Senchenkova, E.Y.; Russell, J.; Almeida-Paula, L.D.; Harding, J.W.; Granger, D.N. Angiotensin II-mediated microvascular thrombosis. Hypertension 2010, 56, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Mogielnicki, A.; Chabielska, E.; Pawlak, R.; Szemraj, J.; Buczko, W. Angiotensin II enhances thrombosis development in renovascular hypertensive rats. Thromb. Haemost. 2005, 93, 1069–1076. [Google Scholar] [CrossRef]
- Ishikawa, M.; Sekizuka, E.; Yamaguchi, N.; Nakadate, H.; Terao, S.; Granger, D.N.; Minamitani, H. Angiotensin II type 1 receptor signaling contributes to platelet-leukocyte-endothelial cell interactions in the cerebral microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2306–H2315. [Google Scholar] [CrossRef]
- Lip, G.Y. Hypertension and the prothrombotic state. J. Hum. Hypertens. 2000, 14, 687–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remkova, A.; Remko, M. The role of renin-angiotensin system in prothrombotic state in essential hypertension. Physiol. Res. 2010, 59, 13–23. [Google Scholar] [PubMed]
- Morishita, R.; Gibbons, G.H.; Ellison, K.E.; Lee, W.; Zhang, L.; Yu, H.; Kaneda, Y.; Ogihara, T.; Dzau, V.J. Evidence for direct local effect of angiotensin in vascular hypertrophy. In vivo gene transfer of angiotensin converting enzyme. J. Clin. Investig. 1994, 94, 978–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankey, G.J.; Heart Outcomes Prevention Evaluation; Perindopril Protection Against Recurrent Stroke Study; Losartan Intervention for Endpoint Reduction in Hypertension Study. Angiotensin-converting enzyme inhibitors for stroke prevention: Is there HOPE for PROGRESS After LIFE? Stroke 2003, 34, 354–356. [Google Scholar] [CrossRef] [Green Version]
- Nadar, S.; Lip, G.Y. The prothrombotic state in hypertension and the effects of antihypertensive treatment. Curr. Pharm. Des. 2003, 9, 1715–1732. [Google Scholar] [CrossRef]
- Celi, A.; Cianchetti, S.; Dell’Omo, G.; Pedrinelli, R. Angiotensin II, tissue factor and the thrombotic paradox of hypertension. Expert Rev. Cardiovasc. Ther. 2010, 8, 1723–1729. [Google Scholar] [CrossRef]
- Matsumoto, T.; Horie, M. Angiotensin-converting enzyme inhibition and fibrinolytic balance. Hypertens. Res. 2011, 34, 448–449. [Google Scholar] [CrossRef]
- Hernandez Prada, J.A.; Ferreira, A.J.; Katovich, M.J.; Shenoy, V.; Qi, Y.; Santos, R.A.; Castellano, R.K.; Lampkins, A.J.; Gubala, V.; Ostrov, D.A.; et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension 2008, 51, 1312–1317. [Google Scholar] [CrossRef] [Green Version]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef]
- Kucharewicz, I.; Pawlak, R.; Matys, T.; Pawlak, D.; Buczko, W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7). Hypertension 2002, 40, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory Dis.tress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Varagic, J.; Ahmad, S.; Nagata, S.; Ferrario, C.M. ACE2: Angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr. Hypertens. Rep. 2014, 16, 420. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Trask, A.J.; Averill, D.B.; Ganten, D.; Chappell, M.C.; Ferrario, C.M. Primary role of angiotensin-converting enzyme-2 in cardiac production of angiotensin-(1-7) in transgenic Ren-2 hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3019–H3024. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Kassiri, Z.; Patel, M.P.; Chappell, M.; Butany, J.; Backx, P.H.; Tsushima, R.G.; Scholey, J.W.; Khokha, R.; Penninger, J.M. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc. Res. 2007, 75, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Bodiga, S.; Fan, D.; Das, S.K.; Wang, Z.; Wang, W.; Basu, R.; Zhong, J.; Kassiri, Z.; Oudit, G.Y. Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1-7 in experimental heart failure in angiotensin-converting enzyme 2-null mice. Hypertension 2012, 59, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Lely, A.T.; Hamming, I.; van Goor, H.; Navis, G.J. Renal ACE2 expression in human kidney Disease. J. Pathol. 2004, 204, 587–593. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, X.R.; Chen, H.Y.; Penninger, J.M.; Lan, H.Y. Loss of angiotensin-converting enzyme 2 enhances TGF-beta/Smad-mediated renal fibrosis and NF-kappaB-driven renal inflammation in a mouse model of obstructive nephropathy. Lab. Investig. 2012, 92, 650–661. [Google Scholar] [CrossRef]
- Wong, D.W.; Oudit, G.Y.; Reich, H.; Kassiri, Z.; Zhou, J.; Liu, Q.C.; Backx, P.H.; Penninger, J.M.; Herzenberg, A.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am. J. Pathol. 2007, 171, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Kowalczuk, S.; Broer, A.; Tietze, N.; Vanslambrouck, J.M.; Rasko, J.E.; Broer, S. A protein complex in the brush-border membrane explains a Hartnup Dis.order allele. FASEB J. 2008, 22, 2880–2887. [Google Scholar] [CrossRef] [PubMed]
- Camargo, S.M.; Singer, D.; Makrides, V.; Huggel, K.; Pos, K.M.; Wagner, C.A.; Kuba, K.; Danilczyk, U.; Skovby, F.; Kleta, R.; et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 2009, 136, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarajapu, Y.P.; Bhatwadekar, A.D.; Caballero, S.; Hazra, S.; Shenoy, V.; Medina, R.; Kent, D.; Stitt, A.W.; Thut, C.; Finney, E.M.; et al. Activation of the ACE2/angiotensin-(1-7)/Mas receptor axis enhances the reparative function of dysfunctional diabetic endothelial progenitors. Diabetes 2013, 62, 1258–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Micro Biol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Hu, B.; Wang, B.; Wang, M.N.; Zhang, Q.; Zhang, W.; Wu, L.J.; Ge, X.Y.; Zhang, Y.Z.; Daszak, P.; et al. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2015, 90, 3253–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchdoerfer, R.N.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.L.; Cottrell, C.A.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 15701. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. BioChem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaimes, J.A.; Andre, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Dis.tinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Deshotels, M.R.; Xia, H.; Sriramula, S.; Lazartigues, E.; Filipeanu, C.M. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an angiotensin II type I receptor-dependent mechanism. Hypertension 2014, 64, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.P.; Lim, W.K.; Velkoska, E.; van Gaal, W.J.; Ryan, J.E.; Savige, J.; Burrell, L.M. N-terminal pro-brain natriuretic peptide and angiotensin-converting enzyme-2 levels and their association with postoperative cardiac complications after emergency orthopedic surgery. Am. J. Cardiol. 2012, 109, 1365–1373. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and bioChem.ical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, D.; Wysocki, J.; Satchell, K. Soluble angiotensin-converting enzyme 2: A potential approach for coronavirus infection therapy? Clin. Sci. (Lond) 2020, 134, 543–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Akhmerov, A.; Marban, E. COVID-19 and the Heart. Circ. Res. 2020, 126, 1443–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hammerle, M.; Esk, C.; Bagley, J.A.; et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020. [Google Scholar] [CrossRef]
- Goldsmith, C.S.; Miller, S.E.; Martines, R.B.; Bullock, H.A.; Zaki, S.R. Electron microscopy of SARS-CoV-2: A challenging task. Lancet 2020, 395, e99. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China. JAMA 2020, 323, 1239. [Google Scholar] [CrossRef]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memish, Z.A.; Al-Tawfiq, J.A.; Makhdoom, H.Q.; Assiri, A.; Alhakeem, R.F.; Albarrak, A.; Alsubaie, S.; Al-Rabeeah, A.A.; Hajomar, W.H.; Hussain, R.; et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J. Infect. Dis. 2014, 210, 1590–1594. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhou, P.; Wei, Y.; Yue, H.; Wang, Y.; Hu, M.; Zhang, S.; Cao, T.; Yang, C.; Li, M.; et al. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, 172, 629–632. [Google Scholar] [CrossRef] [Green Version]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a large series of COVID-19 cases from Northern Italy. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Zhang, W.; Du, R.H.; Li, B.; Zheng, X.S.; Yang, X.L.; Hu, B.; Wang, Y.Y.; Xiao, G.F.; Yan, B.; Shi, Z.L.; et al. Molecular and serological investigation of 2019-nCoV infected patients: Implication of multiple shedding routes. Emerg. Microbes. Infect. 2020, 9, 386–389. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Tong, J.; Liu, M.; Shen, Y.; Guo, D. Evaluation of coronavirus in tears and conjunctival secretions of patients with SARS-CoV-2 infection. J. Med. Virol. 2020, 92, 589–594. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Duan, C.; Zeng, Y.; Tong, Y.; Nie, Y.; Yang, Y.; Chen, Z.; Chen, C. Ocular Findings and Proportion with Conjunctival SARS-COV-2 in COVID-19 Patients. Ophthalmology 2020, 127, 982–983. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Chen, L.; Deng, C.; Zou, X.; Liu, W.; Yu, H.; Chen, B.; Sun, X. The evidence of SARS-CoV-2 infection on ocular surface. Ocul. Surf. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, A.; Li, H.; Zheng, K.; Zhuang, Z.; Chen, Z.; Shi, Y.; Zhang, Z.; Chen, S.-B.; Liu, X.; et al. Isolation of infectious SARS-CoV-2 from urine of a COVID-19 patient. Emerg. Microbes Infect. 2020, 9, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, L.; Deng, Q.; Zhang, G.; Wu, K.; Ni M.D., L.; Yang, Y.; Liu, B.; Wang, W.; Wei, C.; et al. The presence of SARS-CoV-2 RNA in the feces of COVID-19 patients. J. Med. Virol. 2020, 92, 833–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Y.; Xu, S.B.; Lin, Y.X.; Tian, D.; Zhu, Z.Q.; Dai, F.H.; Wu, F.; Song, Z.G.; Huang, W.; Chen, J.; et al. Persistence and clearance of viral RNA in 2019 novel coronavirus Disease rehabilitation patients. Chin. Med. J. 2020, 133, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Dreher, M.; Kersten, A.; Bickenbach, J.; Balfanz, P.; Hartmann, B.; Cornelissen, C.; Daher, A.; Stöhr, R.; Kleines, M.; Lemmen, S.W.; et al. The characteristics of 50 hospitalized COVID-19 patients with and without ARDS. Dtsch. Aerzteblatt Online 2020, 117, 271–278. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients with COVID-19. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Chen, y.; Feng, Z.; Diao, B.; Wang, R.; Wang, G.; Wang, C.; Tan, Y.; Liu, L.; Wang, C.; Liu, Y.; et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. medRxiv 2020. [Google Scholar] [CrossRef]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ Dis.tribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology 2020. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; Al Rasheed, M.; et al. Pathophysiology of SARS-CoV-2: Targeting of endothelial cells renders a complex Disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. medRxiv 2020. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schett, G.; Sticherling, M.; Neurath, M.F. COVID-19: Risk for cytokine targeting in chronic inflammatory Diseases? Nat. Rev. Immunol. 2020, 20, 271–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 2145. [Google Scholar] [CrossRef]
- Moore, J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.E.; Chong, V.C.L.; Chan, S.S.W.; Lim, G.H.; Lim, K.G.E.; Tan, G.B.; Mucheli, S.S.; Kuperan, P.; Ong, K.H. Hematologic parameters in patients with COVID-19 infection. Am. J. Hematol. 2020, 95, E131–E134. [Google Scholar] [CrossRef] [Green Version]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef]
- Marone, E.M.; Rinaldi, L.F. Upsurge of deep venous thrombosis in patients affected by COVID-19: Preliminary data and possible explanations. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 694–695. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Der Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.; Stirling, D.; Ludlam, C.A. Interleukin 6 and haemostasis. Br. J. Haematol. 2001, 115, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Stouthard, J.M.; Levi, M.; Hack, C.E.; Veenhof, C.H.; Romijn, H.A.; Sauerwein, H.P.; van der Poll, T. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb. Haemost. 1996, 76, 738–742. [Google Scholar] [CrossRef]
- Stirling, D.; Hannant, W.A.; Ludlam, C.A. Transcriptional activation of the factor VIII gene in liver cell lines by interleukin-6. Thromb. Haemost. 1998, 79, 74–78. [Google Scholar]
- Burstein, S.A.; Peng, J.; Friese, P.; Wolf, R.F.; Harrison, P.; Downs, T.; Hamilton, K.; Comp, P.; Dale, G.L. Cytokine-induced alteration of platelet and hemostatic function. Stem Cells 1996, 14, 154–162. [Google Scholar] [CrossRef]
- Ishibashi, T.; Kimura, H.; Shikama, Y.; Uchida, T.; Kariyone, S.; Hirano, T.; Kishimoto, T.; Takatsuki, F.; Akiyama, Y. Interleukin-6 is a potent thrombopoietic factor in vivo in mice. Blood 1989, 74, 1241–1244. [Google Scholar] [CrossRef] [Green Version]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Narazaki, M.; Kishimoto, T.; Tosato, G. Receptor engagement by viral interleukin-6 encoded by Kaposi sarcoma-associated herpesvirus. Blood 2001, 98, 3042–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espigol-Frigole, G.; Planas-Rigol, E.; Ohnuki, H.; Salvucci, O.; Kwak, H.; Ravichandran, S.; Luke, B.; Cid, M.C.; Tosato, G. Identification of IL-23p19 as an endothelial proinflammatory peptide that promotes gp130-STAT3 signaling. Sci. Signal. 2016, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, G.M.; Green, D.; Bellmeyer, A.; Baker, C.M.; Burgess, Z.; Rajamannan, N.; Christman, J.W.; Foiles, N.; Kamp, D.W.; Ghio, A.J.; et al. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J. Clin. Investig. 2007, 117, 2952–2961. [Google Scholar] [CrossRef] [Green Version]
- Bennet, A.M.; Prince, J.A.; Fei, G.Z.; Lyrenas, L.; Huang, Y.; Wiman, B.; Frostegard, J.; Faire, U. Interleukin-6 serum levels and genotypes influence the risk for myocardial infarction. Atherosclerosis 2003, 171, 359–367. [Google Scholar] [CrossRef]
- Stern, D.; Nawroth, P.; Handley, D.; Kisiel, W. An endothelial cell-dependent pathway of coagulation. Proc. Natl. Acad. Sci. USA 1985, 82, 2523–2527. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Agstam, S.; Vijay, J.; Gupta, A.; Bansal, S. Acute Pulmonary Embolism: An unseen villain in COVID-19. Indian Hear. J. 2020. [Google Scholar] [CrossRef]
- Palmieri, L.; Vanacore, N.; Donfrancesco, C.; Lo Noce, C.; Canevelli, M.; Punzo, O.; Raparelli, V.; Pezzotti, P.; Riccardo, F.; Bella, A.; et al. Clinical Characteristics of Hospitalized Individuals Dying with COVID-19 by Age Group in Italy. J. Gerontol. A Biol. Sci. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Bhatraju, P.K.; Ghassemieh, B.J.; Nichols, M.; Kim, R.; Jerome, K.R.; Nalla, A.K.; Greninger, A.L.; Pipavath, S.; Wurfel, M.M.; Evans, L.; et al. Covid-19 in Critically Ill Patients in the Seattle Region-Case Series. N. Engl. J. Med. 2020, 382, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 198. [Google Scholar] [CrossRef] [PubMed]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 with Fatal Outcome: Results From a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory Dis.tress syndrome. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Cholankeril, G.; Podboy, A.; Aivaliotis, V.I.; Tarlow, B.; Pham, E.A.; Spencer, S.; Kim, D.; Hsing, A.; Ahmed, A. High Prevalence of Concurrent Gastrointestinal Manifestations in Patients with SARS-CoV-2: Early Experience from California. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Redd, W.D.; Zhou, J.C.; Hathorn, K.E.; McCarty, T.R.; Bazarbashi, A.N.; Thompson, C.C.; Shen, L.; Chan, W.W. Prevalence and Characteristics of Gastrointestinal Symptoms in Patients with SARS-CoV-2 Infection in the United States: A Multicenter Cohort Study. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Liang, T.J. Is SARS-CoV-2 Also an Enteric Pathogen with Potential Fecal-Oral Transmission: A COVID-19 Virological and Clinical Review. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Bhayana, R.; Som, A.; Li, M.D.; Carey, D.E.; Anderson, M.A.; Blake, M.A.; Catalano, O.; Gee, M.S.; Hahn, P.F.; Harisinghani, M.; et al. Abdominal Imaging Findings in COVID-19: Preliminary Observations. Radiology 2020. [Google Scholar] [CrossRef] [PubMed]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, 60. [Google Scholar] [CrossRef]
- Berekashvili, K.; Dmytriw, A.A.; Vulkanov, V.; Agarwal, S.; Khaneja, A.; Turkel-Parella, D.; Liff, J.; Farkas, J.; Nandakumar, T.; Zhou, T.; et al. Etiologic Subtypes of IsChem.ic Stroke in SARS-COV-2 Virus patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Benussi, A.; Pilotto, A.; Premi, E.; Libri, I.; Giunta, M.; Agosti, C.; Alberici, A.; Baldelli, E.; Benini, M.; Bonacina, S.; et al. Clinical characteristics and outcomes of inpatients with neurological Disease and COVID-19. Neurology 2020. [Google Scholar] [CrossRef] [PubMed]
- de Havenon, A.; Ney, J.; Callaghan, B.; Delic, A.; Hohmann, S.; Shippey, E.; Yaghi, S.; Anadani, M.; Esper, G.; Majersik, J. A Rapid Decrease in Stroke, Acute Coronary Syndrome, and Corresponding Interventions at 65 United States Hospitals Following Emergence of COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Mao, L.; Wang, M.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; Li, Y.; Jin, H.; et al. Neurological Manifestations of Hospitalized Patients with COVID-19 in Wuhan, China: A retrospective case series study. medRxiv 2020. [Google Scholar] [CrossRef]
- Coolen, T.; Lolli, V.; Sadeghi, N.; Rovai, A.; Trotta, N.; Taccone, F.S.; Creteur, J.; Henrard, S.; Goffard, J.-C.; Dewitte, O.; et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology 2020. [Google Scholar] [CrossRef]
- De Rosa, S.; Spaccarotella, C.; Basso, C.; Calabro, M.P.; Curcio, A.; Filardi, P.P.; Mancone, M.; Mercuro, G.; Muscoli, S.; Nodari, S.; et al. Reduction of hospitalizations for myocardial infarction in Italy in the COVID-19 era. Eur. Heart J. 2020. [Google Scholar] [CrossRef]
- Garcia, S.; Albaghdadi, M.S.; Meraj, P.M.; Schmidt, C.; Garberich, R.; Jaffer, F.A.; Dixon, S.; Rade, J.J.; Tannenbaum, M.; Chambers, J.; et al. Reduction in ST-Segment Elevation Cardiac Catheterization Laboratory Activations in the United States during COVID-19 Pandemic. J. Am. Coll Cardiol. 2020. [Google Scholar] [CrossRef]
- De Filippo, O.; D’Ascenzo, F.; Angelini, F.; Bocchino, P.P.; Conrotto, F.; Saglietto, A.; Secco, G.G.; Campo, G.; Gallone, G.; Verardi, R.; et al. Reduced Rate of Hospital Admissions for ACS during Covid-19 Outbreak in Northern Italy. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Solomon, M.D.; McNulty, E.J.; Rana, J.S.; Leong, T.K.; Lee, C.; Sung, S.H.; Ambrosy, A.P.; Sidney, S.; Go, A.S. The Covid-19 Pandemic and the Incidence of Acute Myocardial Infarction. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease. Circulation 2020. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.C.; Kim, J.Y.; Kim, H.A.; Han, S. COVID-19-related myocarditis in a 21-year-old female patient. Eur Heart J. 2020, 41, 1859. [Google Scholar] [CrossRef] [PubMed]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, S.; Peretto, G.; Gramegna, M.; Palmisano, A.; Villatore, A.; Vignale, D.; De Cobelli, F.; Tresoldi, M.; Cappelletti, A.M.; Basso, C.; et al. Acute myocarditis presenting as a reverse Tako-Tsubo syndrome in a patient with SARS-CoV-2 respiratory infection. Eur. Heart J. 2020, 41, 1861–1862. [Google Scholar] [CrossRef]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential Effects of Coronaviruses on the Cardiovascular System: A Review. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper, L.T., Jr.; Chahal, C.A.A. Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory Dis.tress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Colon, C.M.; Barrios, J.G.; Chiles, J.W.; McElwee, S.K.; Russell, D.W.; Maddox, W.R.; Kay, G.N. Atrial Arrhythmias in COVID-19 Patients. JACC Clin. Electrophysiol. 2020. [Google Scholar] [CrossRef]
- Kim, M.A.; Yang, D.; Kida, K.; Molotkova, N.; Yeo, S.J.; Varki, N.; Iwata, M.; Dalton, N.D.; Peterson, K.L.; Siems, W.E.; et al. Effects of ACE2 inhibition in the post-myocardial infarction heart. J. Card. Fail. 2010, 16, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Merwe, L.; Cloete, R.; Revera, M.; Heradien, M.; Goosen, A.; Corfield, V.A.; Brink, P.A.; Moolman-Smook, J.C. Genetic variation in angiotensin-converting enzyme 2 gene is associated with extent of left ventricular hypertrophy in hypertrophic cardiomyopathy. Hum. Genet. 2008, 124, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Galvan Casas, C.; Catala, A.; Carretero Hernandez, G.; Rodriguez-Jimenez, P.; Fernandez Nieto, D.; Rodriguez-Villa Lario, A.; Navarro Fernandez, I.; Ruiz-Villaverde, R.; Falkenhain, D.; Llamas Velasco, M.; et al. Classification of the cutaneous manifestations of COVID-19: A rapid prospective nationwide consensus study in Spain with 375 cases. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Bouaziz, J.D.; Duong, T.; Jachiet, M.; Velter, C.; Lestang, P.; Cassius, C.; Arsouze, A.; Domergue Than Trong, E.; Bagot, M.; Begon, E.; et al. Vascular skin symptoms in COVID-19: A french observational study. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef]
- Piccolo, V.; Neri, I.; Filippeschi, C.; Oranges, T.; Argenziano, G.; Battarra, V.C.; Berti, S.; Manunza, F.; Fortina, A.B.; Di Lernia, V.; et al. Chilblain-like lesions during COVID-19 epidemic: A preliminary study on 63 patients. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef]
- Locatelli, A.G.; Robustelli Test, E.; Vezzoli, P.; Carugno, A.; Moggio, E.; Consonni, L.; Gianatti, A.; Sena, P. Histologic features of long lasting chilblain-like lesions in a pediatric COVID-19 patient. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like Disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020. [Google Scholar] [CrossRef]
- Toubiana, J.; Poirault, C.; Corsia, A.; Bajolle, F.; Fourgeaud, J.; Angoulvant, F.; Debray, A.; Basmaci, R.; Salvador, E.; Biscardi, S.; et al. Outbreak of Kawasaki Disease in children during COVID-19 pandemic: A prospective observational study in Paris, France. BMJ 2020. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; Lopez, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; Francois, B.; Seve, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- South, A.M.; Tomlinson, L.; Edmonston, D.; Hiremath, S.; Sparks, M.A. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 2020, 16, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Fuster, J.J.; Walsh, K. The good, the bad, and the ugly of interleukin-6 signaling. EMBO J. 2014, 33, 1425–1427. [Google Scholar] [CrossRef] [Green Version]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Bronneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Fischer, C.P. Physiological roles of muscle-derived interleukin-6 in response to exercise. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 265–271. [Google Scholar] [CrossRef]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; Debuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020. [Google Scholar] [CrossRef]
- Kim, M.; Allen, B.; Korhonen, E.A.; Nitschke, M.; Yang, H.W.; Baluk, P.; Saharinen, P.; Alitalo, K.; Daly, C.; Thurston, G.; et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J. Clin. Investig. 2016, 126, 3511–3525. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| System | Finding |
|---|---|
| Pulmonary | embolism |
| microthrombi | |
| parenchymal infarcts | |
| intussusceptive neovascularization | |
| Gastrointestinal | bowel ischemia/infarction |
| thrombosis of portal/sinusoidal vessels occasional arterial thrombi | |
| hepatic hemorrhages/necrosis | |
| Central nervous system | stroke, transient ischemic attack |
| subcortical bleeds | |
| microthrombi, ischemic infarcts | |
| Cardiovascular | cardiomyopathy |
| venous thromboembolism | |
| Skin | erythema pernio-like lesions, i.e., “pseudo chilblains” |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labò, N.; Ohnuki, H.; Tosato, G. Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection. Cells 2020, 9, 1583. https://doi.org/10.3390/cells9071583
Labò N, Ohnuki H, Tosato G. Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection. Cells. 2020; 9(7):1583. https://doi.org/10.3390/cells9071583
Chicago/Turabian StyleLabò, Nazzarena, Hidetaka Ohnuki, and Giovanna Tosato. 2020. "Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection" Cells 9, no. 7: 1583. https://doi.org/10.3390/cells9071583
APA StyleLabò, N., Ohnuki, H., & Tosato, G. (2020). Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection. Cells, 9(7), 1583. https://doi.org/10.3390/cells9071583