Canonical and Noncanonical Autophagy as Potential Targets for COVID-19

 , and
, and 
Abstract
:1. No Drug for this Bug (and Many Others) Yet
2. Studies on SARS-CoV-2’s Counterparts Might Reveal Therapeutic Targets
3. Autophagy Interplays with the Replication Cycles of Multiple Virus Groups
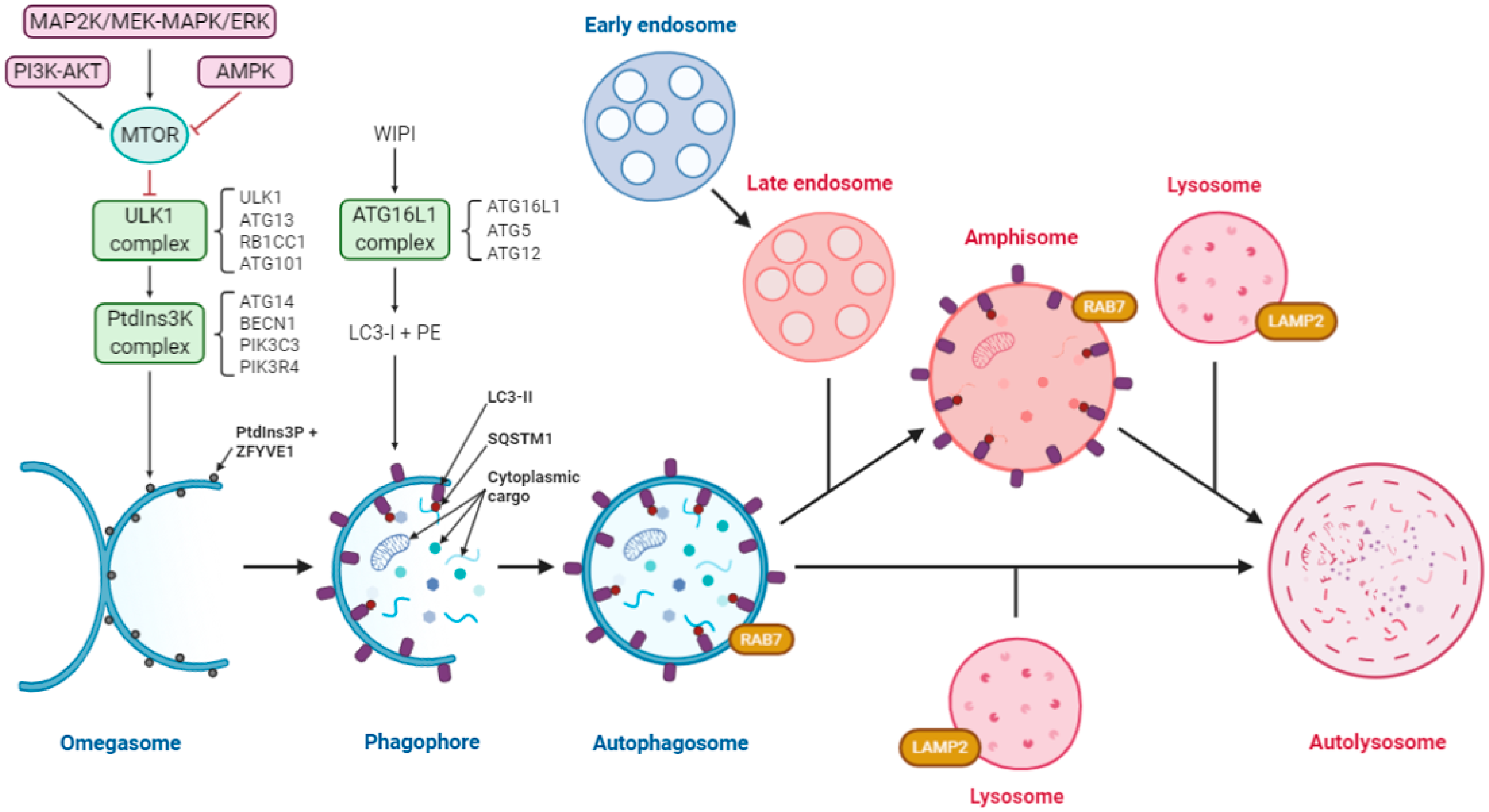
4. Both Autophagy and Coronavirus Induce the Formation of Analogous Vesicular Structures
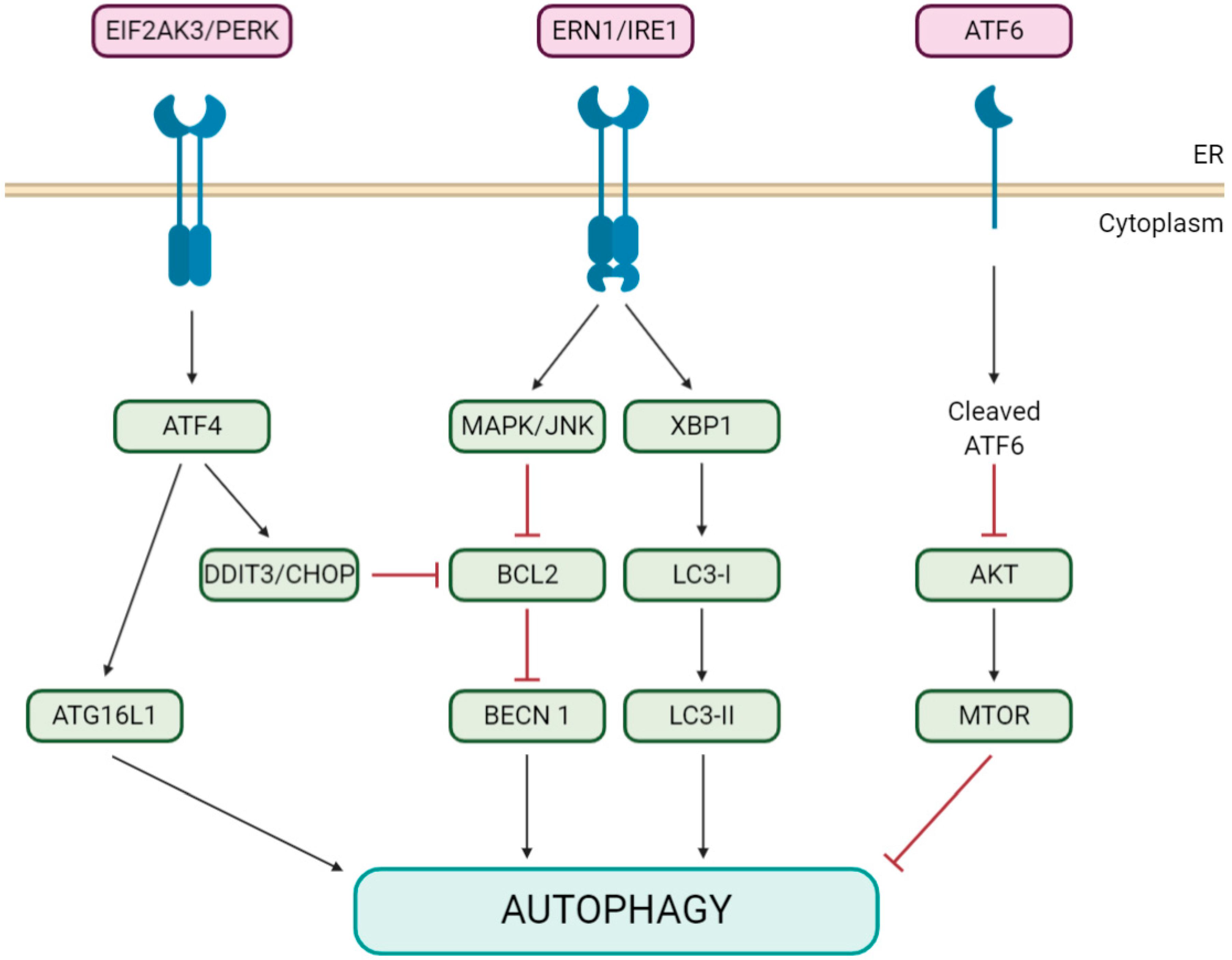
5. Coronaviruses Appear to Modulate Pivotal Initiators of Both Autophagy and Apoptosis
6. Coronavirus Corrupt and Block Autophagy via NSP6 and Some Accessory Proteins
7. Alternative Autophagy Pathways Might be Implicated in Coronavirus Infections
8. Autophagy Modulators are Promising Anticoronavirals

{kind=link}
{kind=link}
| Drug | Action Mechanism on Autophagy | Coronavirus Species | |
|---|---|---|---|
| Inhibited | Non-Inhibited a | ||
| 3-MA | Inhibition of class III PtdIns3K [102] | MHV [51], PEDV [58] | |
| Bafilomycin A1 | Inhibition of V-ATPase, raise lysosomal/vacuolar pH and inhibition of autolysosome formation [24,103] | PEDV [60] | |
| (Hydroxy-) Chloroquine * | Raise lysosomal pH, inhibit autolysosome formation and disorganize Golgi [53] | PEDV [58], SARS-CoV [104,105] SARS-CoV-2 [97,106] | |
| GW5074/Dramafenib * | Inhibition of RAF1/c-Raf1 [107] | MERS-CoV [43] | |
| LY294002 | Inhibitor of PtdIns3K and PI3K [108] | TGEV [57] | |
| Nitazoxanide/Alinia * | Blockage of late-stage lysosome acidification [96] | SARS-CoV-2 [97] | |
| Reserpine * | Inhibitor of autolysosome formation [109] | SARS-CoV [79] | |
| UO126 | Inhibition of MAPK/ERK pathway [73] | MERS-CoV [43] | |
| Wortmannin | Inhibitor of PtdIns3K and PI3Ks [108] | MERS-CoV [43] | IBV [31], PEDV [60], TGEV [57] |
9. Outlook and Challenges
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-ncov and naming it sars-cov-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. International Health Regulations (2005); World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Reperant, L.A.; Osterhaus, A.D. Aids, avian flu, sars, mers, ebola, zika… what next? Vaccine 2017, 35, 4470–4474. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Y.; Zhang, M.; Wang, H.; Zhao, Q.; Liu, J. Updated approaches against sars-cov-2. Antimicrob. Agents Chemother. 2020, 64, e00483-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.; Kok, K.; Zhu, Z.; Chu, H.; To, K.; Yuan, S.; Yuen, K. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient. Emerg. Microbes Infect. 2020, 9, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enjuanes, L.; Zuñiga, S.; Castaño-Rodriguez, C.; Gutierrez-Alvarez, J.; Canton, J.; Sola, I. Molecular basis of coronavirus virulence and vaccine development. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2016; Volume 96, pp. 245–286. [Google Scholar]
- Canton, J.; Fehr, A.R.; Fernandez-Delgado, R.; Gutierrez-Alvarez, F.J.; Sanchez-Aparicio, M.T.; García-Sastre, A.; Perlman, S.; Enjuanes, L.; Sola, I. Mers-cov 4b protein interferes with the nf-κb-dependent innate immune response during infection. PLoS Pathog. 2018, 14, e1006838. [Google Scholar] [CrossRef]
- Taxonomy, I.V. Release. 2019; International Committee on Taxonomy of Viruses: Berlin, Germany, 2019. [Google Scholar]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in china, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-em structure of the 2019-ncov spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ace2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. Sars coronavirus entry into host cells through a novel clathrin-and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J. Characterization of spike glycoprotein of sars-cov-2 on virus entry and its immune cross-reactivity with sars-cov. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hemert, M.J.; van den Worm, S.H.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. Sars-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The atg16l complex specifies the site of lc3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014. [Google Scholar] [CrossRef]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, L. Regulation of atg and autophagy initiation. In Autophagy: Biology and Diseases; Springer: Singapore, 2019; pp. 41–65. [Google Scholar]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and viral infection. In Autophagy Regulation of Innate Immunity; Springer: Singapore, 2019; pp. 55–78. [Google Scholar] [CrossRef]
- Wong, H.H.; Sanyal, S. Manipulation of Autophagy by (+) RNA Viruses; Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Yin, H.-c.; Shao, S.-l.; Jiang, X.-j.; Xie, P.-y.; Sun, W.-s.; Yu, T.-f. Interactions between autophagy and DNA viruses. Viruses 2019, 11, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.E.v.d.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Sars-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 2008, 6, e226. [Google Scholar] [CrossRef]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baliji, S.; Cammer, S.A.; Sobral, B.; Baker, S.C. Detection of nonstructural protein 6 in murine coronavirus-infected cells and analysis of the transmembrane topology by using bioinformatics and molecular approaches. J. Virol. 2009, 83, 6957–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Li, B.; Liu, M.; Zhou, H.; He, K.; Fan, H. Nonstructural protein 6 of porcine epidemic diarrhea virus induces autophagy to promote viral replication via the pi3k/akt/mtor axis. Vet. Microbiol. 2020, 244, 108684. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy 2010, 6, 186–188. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S.; Gallagher, C.M.; Walter, P. Er-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci 2014, 127, 4078–4088. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor ire1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, M.; Hino, S.-i.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. The er stress sensor ire1 and map kinase erk modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology 2019, 533, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. Mtorc1 serves er stress-triggered apoptosis via selective activation of the ire1–jnk pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of chop/gadd153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical mek/erk signaling pathway regulates autophagy via regulating beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [Green Version]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H. Antiviral potential of erk/mapk and pi3k/akt/mtor signaling modulation for middle east respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. Targeting disease through novel pathways of apoptosis and autophagy. Expert Opin. Ther. Targets 2012, 16, 1203–1214. [Google Scholar] [CrossRef]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A. Skp2 attenuates autophagy through beclin1-ubiquitination and its inhibition reduces mers-coronavirus infection. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Zhao, L.; Jha, B.K.; Wu, A.; Elliott, R.; Ziebuhr, J.; Gorbalenya, A.E.; Silverman, R.H.; Weiss, S.R. Antagonism of the interferon-induced oas-rnase l pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 2012, 11, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Thornbrough, J.M.; Jha, B.K.; Yount, B.; Goldstein, S.A.; Li, Y.; Elliott, R.; Sims, A.C.; Baric, R.S.; Silverman, R.H.; Weiss, S.R. Middle east respiratory syndrome coronavirus ns4b protein inhibits host rnase l activation. MBio 2016, 7, e00258-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, A.; Ghosh, P.K.; Banerjee, S.; Gaughan, C.; Silverman, R.H. Rnase l triggers autophagy in response to viral infections. J. Virol. 2012, 86, 11311–11321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, M.A.; Malathi, K. Rnase l induces autophagy via c-jun n-terminal kinase and double-stranded rna-dependent protein kinase signaling pathways. J. Biol. Chem. 2012, 287, 43651–43664. [Google Scholar] [CrossRef] [Green Version]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus nsp6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin a1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, h-4-ii-e cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does Bafilomycin a1 Block the Fusion of Autophagosomes with Lysosomes? Taylor & Francis: Abingdon, UK, 2008; Volume 4, pp. 849–850. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–23. [Google Scholar]
- Guo, L.; Yu, H.; Gu, W.; Luo, X.; Li, R.; Zhang, J.; Xu, Y.; Yang, L.; Shen, N.; Feng, L. Autophagy negatively regulates transmissible gastroenteritis virus replication. Sci. Rep. 2016, 6, 23864. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, M.; Zhang, X.; Tan, X.; Guo, H.; Zeng, W.; Yan, G.; Memon, A.M.; Li, Z.; Zhu, Y. Porcine epidemic diarrhea virus induces autophagy to benefit its replication. Viruses 2017, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses hijack the lc3-i-positive edemosomes, er-derived vesicles exporting short-lived erad regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.; Gu, M.J.; Kim, C.G.; Kye, Y.C.; Lim, Y.; Lee, J.E.; Park, B.-C.; Chu, H.; Han, S.H.; Yun, C.-H. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antivir. Res. 2017, 146, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.; Denison, M.R.; Virgin, I.; Herbert, W. Coronavirus replication does not require the autophagy gene atg5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, E.J.; Van Der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of atg5/atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Grose, C.; Klionsky, D.J. Alternative Autophagy, Brefeldin a and Viral Trafficking Pathways; Taylor & Francis: Abingdon, UK, 2016; Volume 12, pp. 1429–1430. [Google Scholar] [CrossRef] [Green Version]
- Zeghouf, M.; Guibert, B.; Zeeh, J.-C.; Cherfils, J. Arf, sec7 and Brefeldin a: A Model towards the Therapeutic Inhibition of Guanine Nucleotide-Exchange Factors; Portland Press Ltd. Biochem Soc Trans. 2005, 33, 1265–1268. [Google Scholar] [CrossRef] [Green Version]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479, 450–456. [Google Scholar] [CrossRef]
- Yadav, V.; Panganiban, A.T.; Zu Bentrup, K.H.; Voss, T.G. Influenza infection modulates vesicular trafficking and induces golgi complex disruption. Virusdisease 2016, 27, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Molina, S.; Sanz, M.A.; Madan, V.; Ventoso, I.; Castelló, A.; Carrasco, L. Differential inhibition of cellular and sindbis virus translation by brefeldin a. Virology 2007, 363, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, L. Key components of copi and copii machineries are required for chikungunya virus replication. Biochem. Biophys. Res. Commun. 2017, 493, 1190–1196. [Google Scholar] [CrossRef]
- Laniosz, V.; Dabydeen, S.A.; Havens, M.A.; Meneses, P.I. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. J. Virol. 2009, 83, 8221–8232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus rna replication depends on gbf1-mediated arf1 activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Arakawa, S.; Nishida, Y. Autophagy takes an alternative pathway. Autophagy 2010, 6, 290–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-h.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Mariño, G.; Cianfanelli, V.; Ben-Younès, A.; Troncoso, R.; Markaki, M.; Sica, V. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef]
- Riederer, M.A.; Soldati, T.; Shapiro, A.D.; Lin, J.; Pfeffer, S.R. Lysosome biogenesis requires rab9 function and receptor recycling from endosomes to the trans-golgi network. J. Cell Biol. 1994, 125, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Mou, C.; Yang, X.; Lin, J.; Yang, Q. Mitophagy in tgev infection counteracts oxidative stress and apoptosis. Oncotarget 2016, 7, 27122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, S.A.; Orhon, I.; Morselli, E.; Criollo, A.; Shen, S.; Marino, G.; BenYounes, A.; Benit, P.; Rustin, P.; Maiuri, M.C. Bh3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011, 30, 3918–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Yu, W.; Liu, B.; Wang, Y.; Wang, J.; Xia, K.; Liang, C.; Fang, W.; Zhou, C.; Tao, H. Escin induces caspase-dependent apoptosis and autophagy through the ros/p38 mapk signalling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2017, 8, e3113. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Jan, J.-T.; Ma, S.-H.; Kuo, C.-J.; Juan, H.-F.; Cheng, Y.-S.E.; Hsu, H.-H.; Huang, H.-C.; Wu, D.; Brik, A. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [Green Version]
- Martinet, W.; Verheye, S.; De Meyer, G.R. Everolimus-induced mtor inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy 2007, 3, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.E.; Bay, B.-H.; Wenk, M.R.; Shen, H.-M. Induction of autophagy by palmitic acid via protein kinase c-mediated signaling pathway independent of mtor (mammalian target of rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Q.; Chen, H.-N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N. Ivermectin induces cytostatic autophagy by blocking the pak1/akt axis in breast cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [Green Version]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The fda-approved drug ivermectin inhibits the replication of sars-cov-2 in vitro. Antivir. Res. 2020, 104787. [Google Scholar] [CrossRef]
- Wang, J.; Ren, X.-r.; Piao, H.; Zhao, S.; Osada, T.; Premont, R.T.; Mook, R.A.; Morse, M.A.; Lyerly, H.K.; Chen, W. Niclosamide-induced wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem. J. 2019, 476, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Dieter, P.; Fitzke, E. Ro 31-8220 and ro 31-7549 show improved selectivity for protein kinase c over staurosporine in macrophages. Biochem. Biophys. Res. Commun. 1991, 181, 396–401. [Google Scholar] [CrossRef]
- Jiang, H.; Cheng, D.; Liu, W.; Peng, J.; Feng, J. Protein kinase c inhibits autophagy and phosphorylates lc3. Biochem. Biophys. Res. Commun. 2010, 395, 471–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, S.; Pereira, G.J.; Palmeira-dos-Santos, C.; Calgarotto, A.K.; Martínez-Lacaci, I.; Ferragut, J.A.; Smaili, S.S.; Bincoletto, C. Autophagy regulates selumetinib (azd6244) induced-apoptosis in colorectal cancer cells. Eur. J. Med. Chem. 2016, 122, 611–618. [Google Scholar] [CrossRef]
- Rico-Bautista, E.; Yang, C.-C.; Lu, L.; Roth, G.P.; Wolf, D.A. Chemical genetics approach to restoring p27kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC Biol. 2010, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M. Bay 43-9006 exhibits broad spectrum oral antitumor activity and targets the raf/mek/erk pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-F.; Chen, H.-L.; Tai, W.-T.; Feng, W.-C.; Hsu, C.-H.; Chen, P.-J.; Cheng, A.-L. Activation of phosphatidylinositol 3-kinase/akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T. Protective autophagy elicited by raf→ mek→ erk inhibition suggests a treatment strategy for ras-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Klein, B.; Wörndl, K.; Lütz-Meindl, U.; Kerschbaum, H.H. Perturbation of intracellular k+ homeostasis with valinomycin promotes cell death by mitochondrial swelling and autophagic processes. Apoptosis 2011, 16, 1101. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored snare syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.-P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. Ph-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through dc-sign. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Cell entry by enveloped viruses: Redox considerations for hiv and sars-coronavirus. Antioxid. Redox Signal. 2007, 9, 1009–1034. [Google Scholar]
- Wang, X.; Shen, C.; Liu, Z.; Peng, F.; Chen, X.; Yang, G.; Zhang, D.; Yin, Z.; Ma, J.; Zheng, Z. Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ing1-induced cell cycle arrest in glioblastoma. Cell Death Dis. 2018, 9, 1032. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-ncov) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Shou, J.; Wang, M.; Cheng, X.; Wang, X.; Zhang, L.; Liu, Y.; Fei, C.; Wang, C.; Gu, F.; Xue, F. Tizoxanide induces autophagy by inhibiting pi3k/akt/mtor pathway in raw264. 7 macrophage cells. Arch. Pharmacal Res. 2020, 43, 257–270. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-ncov/sars-cov-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, M.; De Giglio, M.; Roviello, G. Sars cov-2: Recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Dethloff, F.; Emanuel, J.; Weckmann, K.; Heinz, D.E.; Heinemann, N.; Lennarz, M.; Richter, A. Analysis of sars-cov-2-controlled autophagy reveals spermidine, mk-2206, and niclosamide as putative antiviral therapeutics. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Seglen, P.O.; Gordon, P.B. 3-methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome–lysosome fusion is independent of v-atpase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [Green Version]
- Keyaerts, E.; Vijgen, L.; Maes, P.; Neyts, J.; Van Ranst, M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 2004, 323, 264–268. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of sars coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (sars-cov-2). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wu, Y.; Tashiro, S.-i.; Onodera, S.; Ikejima, T. Involvement of pkc signal pathways in oridonin-induced autophagy in hela cells: A protective mechanism against apoptosis. Biochem. Biophys. Res. Commun. 2009, 378, 273–278. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and ly294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.I.; Kim, M.J.; Koh, H.; Lee, J.I.; Namkoong, S.; Oh, W.K.; Park, J. The anti-hypertensive drug reserpine induces neuronal cell death through inhibition of autophagic flux. Biochem. Biophys. Res. Commun. 2015, 462, 402–408. [Google Scholar] [CrossRef]
- Yang, N.; Shen, H.-M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in covid-19. Int. J. Biol. Sci. 2020, 16, 1724. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.; Aivazian, D. Targeting rab gtpases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004, 5, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab gtpases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Andreani, J.; Le Bideau, M.; Duflot, I.; Jardot, P.; Rolland, C.; Boxberger, M.; Wurtz, N.; Rolain, J.-M.; Colson, P.; La Scola, B. In vitro testing of combined hydroxychloroquine and azithromycin on sars-cov-2 shows synergistic effect. Microb. Pathog. 2020, 145, 104228. [Google Scholar] [CrossRef] [PubMed]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; Dupont, H.T. Hydroxychloroquine and azithromycin as a treatment of covid-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 105949. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Brown, K.; Shang, S.; Tamayo, M.H.; Hegyi, K.; Grimsey, N.J.; Cusens, D.; Coulter, S.; Cooper, J. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Investig. 2011, 121, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Moriya, S.; Che, X.-F.; Komatsu, S.; Abe, A.; Kawaguchi, T.; Gotoh, A.; Inazu, M.; Tomoda, A.; Miyazawa, K. Macrolide antibiotics block autophagy flux and sensitize to bortezomib via endoplasmic reticulum stress-mediated chop induction in myeloma cells. Int. J. Oncol. 2013, 42, 1541–1550. [Google Scholar] [CrossRef] [Green Version]
- Keyaerts, E.; Li, S.; Vijgen, L.; Rysman, E.; Verbeeck, J.; Van Ranst, M.; Maes, P. Antiviral activity of chloroquine against human coronavirus oc43 infection in newborn mice. Antimicrob. Agents Chemother. 2009, 53, 3416–3421. [Google Scholar] [CrossRef] [Green Version]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G. Observational study of hydroxychloroquine in hospitalized patients with covid-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.H.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Outcomes of hydroxychloroquine usage in united states veterans hospitalized with covid-19. Med 2020. [Google Scholar] [CrossRef]


| Genus (No. Subgenera) | Subgenus a | Species b | Disease and Host |
|---|---|---|---|
| Alphacoronavirus (14) | Duvinacovirus | HCoV-229E | Common cold in humans |
| Tegacovirus | TGEV | Transmissible gastroenteritis disease in pigs | |
| Pedacovirus | PEDV | Porcine epidemic diarrhea disease | |
| Betacoronavirus (5) | Embecovirus | HCoV-OC43 MHV | Common cold in humans Murine hepatitis disease |
| Merbecovirus | MERS-CoV | MERS in humans | |
| Sarbecovirus | SARS-CoV* SARS-CoV-2* | SARS in humans COVID-19 in humans | |
| Deltacoronavirus (3) | Buldecovirus | PDCoV | Acute gastrointestinal disorders in neonatal piglets |
| Gammacoronavirus (3) | Igacovirus | IBV | Infectious bronchitis disease in chickens |
| Virus/NSP6 | Cell Lines | Autophagy Vesicle Marker | Colocalized Viral Protein/Element |
|---|---|---|---|
| IBV | Vero | LC3 | dsRNA [31] |
| WIPI2, ATG5 [31] | |||
| MHV | HEK293 | LC3 | NSP2/3 [59] |
| HeLa | LC3 | NSP2/3 [59] | |
| MEF | LC3 | N, p22, Hel, M [51], NSP2/3 [59] | |
| ATG12 | N [51] | ||
| PEDV | Vero-E6 | LC3 [58] | |
| IPEC-J2 | LC3 | N [60] | |
| SARS-CoV | Vero | LC3 | Replicase proteins [61] |
| TGEV | ST | LC3 [57] | |
| IBV NSP6 | CHO | LC3, SQSTM1/p62 [31] | |
| HEK293 | ATG5, ZFYVE1/DFCP1 [31] | ||
| MEF | LC3 [31] | ||
| Vero | LC3, WIPI2 [50] | ||
| MHV NSP6 | CHO | LC3 [31] | |
| Vero | LC3 [50] | ||
| SARS-CoV NSP6 | CHO | LC3 | NSP6 [31] |
| Gene a | Levels of Viral Infection/Replication b | ||
|---|---|---|---|
| Lower | Equal | Higher | |
| ATG5/Atg5 | MHV [51], PEDV [58] | IBV [31,39], MHV [61] | MERS-CoV [45], TGEV [57] |
| ATG7/Atg7 | MHV [59] | TGEV [57] | |
| BECN1/Becn1 | PEDV [58] | IBV [39] | |
| LC3/Lc3 | MHV [59] | TGEV [57] | |
| Drug | Action Mechanism on Autophagy | Coronavirus Species | |
|---|---|---|---|
| Inhibited | Non-Inhibited a | ||
| ABT-737/Venetoclax * | Release of BECN1 from BCL2 and BCL2L1/Bcl-XL interaction [77] | MERS-CoV [45] | |
| Aescim | Activation of ROS-MAPK/p38 signaling pathway [78] | SARS-CoV [79] | |
| Everolimus/Afinitor * | Inhibition of MTOR [80] | MERS-CoV [43] | |
| GF109203X | Inhibition of PRKC/PKC (protein kinase C) [81] | MERS-CoV [43] | |
| Ivermectin * | Inhibition of PAK1 and subsequent AKT phosphorylation [82] | SARS-CoV-2 [83] | |
| Niclosamide * | Inhibition of MTORC1 and ULK1 activities and induction of LC3B expression [83,84] | MERS-CoV [45] | |
| Rapamycin/ Sirolumus * | Inhibition of MTOR [33] | MERS-CoV [43], MHV [59], TGEV [57], PEDV [60] | PEDV [58] |
| Ro-31-8220 | Inhibition of PRKC/PKC [85,86] | MERS-CoV [43] | |
| Selumetinib * | Inhibitor of MAP2K1/MEK1-MAP2K2/MEK2 [87] | MERS-CoV [43] | |
| SMIP004 | Inhibition of SKP2 [88] | MERS-CoV [45] | |
| Sorafenib/Nexavar * | Inhibition of RAF-MAP2K-MAPK/ERK signaling pathway and VEGF receptor tyrosine kinase [89] and activation of AKT [90] | MERS-CoV [43] | |
| Trametinib/Tafinlar * | Inhibitor of MAP2K1/MEK1-MAP2K2/MEK2 [91] | MERS-CoV [43] | |
| Valinomycin | Electrogenic K+ ionophore that causes loss of the mitochondrial membrane potential and stimulates mitophagy [92] | MERS-CoV [45], SARS-CoV [79] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells 2020, 9, 1619. https://doi.org/10.3390/cells9071619
Bello-Perez M, Sola I, Novoa B, Klionsky DJ, Falco A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells. 2020; 9(7):1619. https://doi.org/10.3390/cells9071619
Chicago/Turabian StyleBello-Perez, Melissa, Isabel Sola, Beatriz Novoa, Daniel J. Klionsky, and Alberto Falco. 2020. "Canonical and Noncanonical Autophagy as Potential Targets for COVID-19" Cells 9, no. 7: 1619. https://doi.org/10.3390/cells9071619
APA StyleBello-Perez, M., Sola, I., Novoa, B., Klionsky, D. J., & Falco, A. (2020). Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells, 9(7), 1619. https://doi.org/10.3390/cells9071619