Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Cell Cultures
2.3. Ngn2-Mediated Neuronal Differentiation of Human iPSCs
2.4. Isolation of Astrocyte- and Neuronal-Derived Extracellular Vesicles from Human Plasma
2.5. Characterization of AEVs and NEVs by Immunoblotting and ELISA
2.6. Neurotoxicity Assays
2.7. Assessment of Neurite Fragmentation by Immunocytochemistry
2.8. Assessment of EV-Mediated Activation of Apoptosis and Necroptosis
2.9. Experimental Design and Statistical Analyses
3. Results
3.1. Characterization of Plasma-Derived EVs
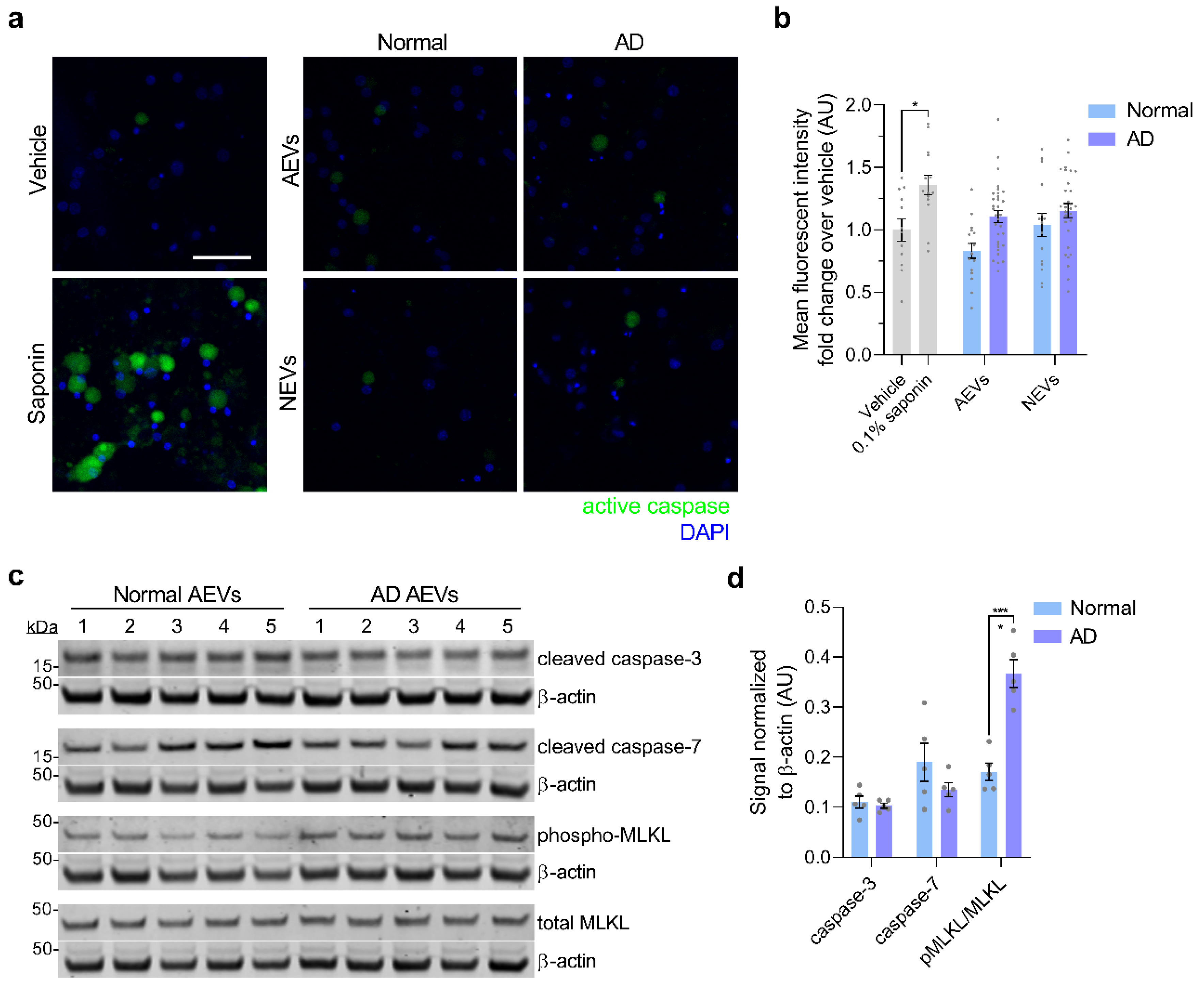
3.2. AD AEVs and NEVs are Neurotoxic
3.3. Neurotoxicity of AD AEVs is Associated with Complement MAC Accumulation, Membrane Disruption, and Necroptosis Activation
3.4. Complement Inhibition Blocks the Neurotoxicity of AD AEVs and NEVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.W. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; I Luebke, J.; Haydar, T.F.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.-A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Farkas, I.; Baranyi, L.; Ishikawa, Y.; Okada, N.; Bohata, C.; Budai, D.; Fukuda, A.; Imai, M.; Okada, H. CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J. Physiol. 2002, 539, 537–545. [Google Scholar] [CrossRef]
- Parsons, E.S.; Stanley, G.J.; Pyne, A.L.B.; Hodel, A.W.; Nievergelt, A.P.; Menny, A.; Yon, A.R.; Rowley, A.; Richter, R.P.; Fantner, G.E.; et al. Single-molecule kinetics of pore assembly by the membrane attack complex. Nat. Commun. 2019, 10, 2066. [Google Scholar] [CrossRef] [Green Version]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.M.; Ager, R.R.; Tenner, A.J.; Noakes, P.G.; Taylor, S.M. The Role of the Complement System and the Activation Fragment C5a in the Central Nervous System. Neuromol. Med. 2009, 12, 179–192. [Google Scholar] [CrossRef]
- Terai, K.; Walker, D.G.; McGeer, E.G.; McGeer, P.L. Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res. 1997, 769, 385–390. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.-A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.I.; Ager, R.R.; Chu, S.-H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123. [Google Scholar] [CrossRef] [Green Version]
- Brachova, L.; Lue, L.-F.; Schultz, J.; El Rashidy, T.; Rogers, J. Association cortex, cerebellum, and serum concentrations of C1q and factor B in Alzheimer’s disease. Mol. Brain Res. 1993, 18, 329–334. [Google Scholar] [CrossRef]
- Fonseca, M.; Kawas, C.H.; Troncoso, J.; Tenner, A.J. Neuronal localization of C1q in preclinical Alzheimer’s disease. Neurobiol. Dis. 2004, 15, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Konishi, Y.; Beach, T.; I Sue, L.; Hampel, H.; Lindholm, K.; Shen, Y. The temporal localization of frame-shift ubiquitin-B and amyloid precursor protein, and complement proteins in the brain of non-demented control patients with increasing Alzheimer’s disease pathology. Neurosci. Lett. 2003, 348, 46–50. [Google Scholar] [CrossRef]
- Webster, S.; Lue, L.-F.; Brachova, L.; Tenner, A.J.; McGeer, P.L.; Terai, K.; Walker, D.G.; Bradt, B.; Cooper, N.R.; Rogers, J. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol. Aging 1997, 18, 415–421. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, M.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Yan, F.; Lin, A.H.-T.; Lambris, J.D.; Alexander, J.J.; Quigg, R.J.; Masliah, E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. USA 2002, 99, 10837–10842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Rajendran, L. The Transcellular Spread of Cytosolic Amyloids, Prions, and Prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging Role of Neuronal Exosomes in the Central Nervous System. Front. Physiol. 2012, 3, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, M.M.; Kaiser, J.; Schwab, M.E. Extracellular Vesicles: Multimodal Envoys in Neural Maintenance and Repair. Trends Neurosci. 2018, 41, 360–372. [Google Scholar] [CrossRef]
- Shi, M.; Sheng, L.; Stewart, T.; Zabetian, C.P.; Zhang, J. New windows into the brain: Central nervous system-derived extracellular vesicles in blood. Prog. Neurobiol. 2019, 175, 96–106. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Mustapic, M.; Eitan, E.; Werner, K.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Front. Mol. Neurosci. 2017, 11, 278. [Google Scholar] [CrossRef] [Green Version]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.; Mustapic, M.; Chia, C.W.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, M.P.; Resnick, S.; Egan, J.M.; et al. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 741–752. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.W.; Dubois, B.; Feldman, H.H.; Fox, N.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Cairns, N.J.; Bigio, E.H.; MacKenzie, I.R.A.; Neumann, M.; Lee, V.M.-Y.; Hatanpaa, K.J.; White, C.L.; Schneider, J.A.; Grinberg, L.T.; Halliday, G.M.; et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Barger, S.; Begley, J.G.; Mark, R.J. Chapter 10 Calcium, Free Radicals, and Excitotoxic Neuronal Death in Primary Cell Culture. Methods Cell Biol. 1995, 46, 187–216. [Google Scholar] [CrossRef]
- Chou, B.-K.; Gu, H.; Gao, Y.; Dowey, S.N.; Wang, Y.; Shi, J.; Li, Y.; Ye, Z.; Cheng, T.; Cheng, L. A Facile Method to Establish Human Induced Pluripotent Stem Cells From Adult Blood Cells Under Feeder-Free and Xeno-Free Culture Conditions: A Clinically Compliant Approach. STEM CELLS Transl. Med. 2015, 4, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.G.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.; Bora, A.; Lässer, C.; Lötvall, J.; Hoen, E.N.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 18389. [Google Scholar] [CrossRef] [PubMed]
- Coumans, F.; Brisson, A.R.; Buzás, E.; Dignat-George, F.; Drees, E.E.; El Andaloussi, S.; Emanueli, C.; Gąsecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; A Jicha, G.; Liu, N.; et al. Extracellular Vesicle-Associated Abeta Mediates Trans-Neuronal Bioenergetic and Ca(2+)-Handling Deficits in Alzheimer’s Disease Models. NPJ Aging Mech. Dis. 2016, 2, 1–11. [Google Scholar] [CrossRef]
- Li, J.; Tang, H.; Zhang, Y.; Tang, C.; Li, B.; Wang, Y.; Gao, Z.; Luo, P.; Yin, A.; Wang, X.; et al. Saponin 1 induces apoptosis and suppresses NF-kappaB-mediated survival signaling in glioblastoma multiforme (GBM). PLoS ONE 2013, 8, e81258. [Google Scholar]
- Wu, C.-A.; Yang, Y.-W. Induction of cell death by saponin and antigen delivery. Pharm. Res. 2004, 21, 271–277. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Mollnes, T.E.; Lea, T.; Harboe, M.; Tschopp, J. Monoclonal Antibodies Recognizing a Neoantigen of Poly(C9) Detect the Human Terminal Complement Complex in Tissue and Plasma. Scand. J. Immunol. 1985, 22, 183–195. [Google Scholar] [CrossRef] [PubMed]
- A Kallio, E.; Lemström, K.; Häyry, P.J.; Ryan, U.S.; Koskinen, P.K. Blockade of Complement Inhibits Obliterative Bronchiolitis in Rat Tracheal Allografts. Am. J. Respir. Crit. Care Med. 2000, 161, 1332–1339. [Google Scholar] [CrossRef]
- Lee, M.K.; Rebhun, L.I.; Frankfurter, A. Posttranslational modification of class III beta-tubulin. Proc. Natl. Acad. Sci. USA 1990, 87, 7195–7199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Lusthaus, M.; Mazkereth, N.; Donin, N.; Fishelson, Z. Receptor-Interacting Protein Kinases 1 and 3, and Mixed Lineage Kinase Domain-Like Protein Are Activated by Sublytic Complement and Participate in Complement-Dependent Cytotoxicity. Front. Immunol. 2018, 9, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Galluzzi, L.; Berghe, T.V.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Boil. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, P.J.; Weidner, A.M.; Levine, H.; Markesbery, W.R.; Murphy, M.P.; Zhang, L.; Dasuri, K.; Fernandez-Kim, S.O.; Bruce-Keller, A.J.; Gavilan, E.; et al. Neuron Specific Toxicity of Oligomeric Amyloid-β: Role for JUN-Kinase and Oxidative Stress. J. Alzheimer’s Dis. 2010, 22, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, C.N.; Aulston, B.; Rockenstein, E.M.; Adame, A.; Prikhodko, O.; Dave, K.N.; Mishra, P.; A Rissman, R.; Yuan, S.H. Neuronal Exosome-Derived Human Tau is Toxic to Recipient Mouse Neurons in vivo. J. Alzheimer’s Dis. 2019, 67, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chopp, M.; Liu, X.S.; Katakowski, M.; Wang, X.; Tian, X.; Wu, D.; Zhang, Z.G. Exosomes Derived from Mesenchymal Stromal Cells Promote Axonal Growth of Cortical Neurons. Mol. Neurobiol. 2016, 54, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Dickens, A.M.; Tovar-y-Romo, L.B.; Yoo, S.-W.; Trout, A.L.; Bae, M.; Kanmogne, M.; Megra, B.; Williams, D.W.; Witwer, K.W.; Gacias, M.; et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal. 2017, 10, eaai7696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front. Mol. Neurosci. 2019, 13, 1208. [Google Scholar] [CrossRef] [Green Version]
- Pulliam, L.; Sun, B.; Mustapic, M.; Chawla, S.; Kapogiannis, D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J. Neurovirol. 2019, 25, 702–709. [Google Scholar] [CrossRef]
- Vedeler, C.; Ulvestad, E.; Bjørge, L.; Conti, G.; Williams, K.; Mørk, S.; Matre, R. The expression of CD59 in normal human nervous tissue. Immunology 1994, 82, 542–547. [Google Scholar]
- Gasque, P.; Fontaine, M.; Morgan, B.P. Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J. Immunol. 1995, 154, 4726–4733. [Google Scholar]
- McGeer, P.; Walker, D.; Akiyama, H.; Kawamata, T.; Guan, A.; Parker, C.; Okada, N.; McGeer, E. Detection of the membrane inhibitor of reactive lysis (CD59) in diseased neurons of Alzheimer brain. Brain Res. 1991, 544, 315–319. [Google Scholar] [CrossRef]
- Yang, L.-B.; Li, R.; Meri, S.; Rogers, J.; Shen, Y. Deficiency of Complement Defense Protein CD59 May Contribute to Neurodegeneration in Alzheimer’s Disease. J. Neurosci. 2000, 20, 7505–7509. [Google Scholar] [CrossRef] [Green Version]
- Yuksel, D.; Oguz, K.K.; Azapagası, E.; Kesici, S.; Cavdarli, B.; Konuskan, B.; Topaloglu, H. Uncontrolled inflammation of the nervous system. Neurol. Clin. Pr. 2018, 8, e18–e20. [Google Scholar] [CrossRef] [PubMed]
- Nevo, Y.; Ben-Zeev, B.; Tabib, A.; Straussberg, R.; Anikster, Y.; Shorer, Z.; Fattal-Valevski, A.; Ta-Shma, A.; Aharoni, S.; Rabie, M.; et al. CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood 2013, 121, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ardicli, D.; Oguz, K.K.; Topaloglu, H.; Taskiran, E.Z.; Kosukcu, C.; Temucin, C.; Haliloglu, G.; Alikasifoglu, M. Neonatal-Onset Recurrent Guillain–Barré Syndrome-Like Disease: Clues for Inherited CD59 Deficiency. Neuropediatrics 2017, 48, 477–481. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G.; Yasojima, K. Alzheimer disease and neuroinflammation. Adv. Dement. Res. 2000, 59, 53–57. [Google Scholar] [CrossRef]
- Sugita, Y.; Masuho, Y. CD59: Its role in complement regulation and potential for therapeutic use. Immunotechnology 1995, 1, 157–168. [Google Scholar] [CrossRef]
- Coelho, D.; Schwartz, S.; Merino, M.M.; Hauert, B.; Topfel, B.; Tieche, C.; Rhiner, C.; Moreno, E. Culling Less Fit Neurons Protects against Amyloid-beta-Induced Brain Damage and Cognitive and Motor Decline. Cell Rep. 2018, 25, 3661–3673. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.Y.F.; Rawji, K.S.; Ghorbani, S.; Xue, M.; Yong, V.W. The benefits of neuroinflammation for the repair of the injured central nervous system. Cell. Mol. Immunol. 2019, 16, 540–546. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Jana, A.; Pahan, K. Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: Implications for Alzheimer’s disease. J. Neurosci. 2010, 30, 12676–12689. [Google Scholar] [CrossRef]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Chang, H.-Y.; Sang, T.-K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; E Spangenberg, E.; Green, K.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Rother, R.P.; A Rollins, S.; Mojcik, C.F.; A Brodsky, R.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Hajishengallis, E.; Kajikawa, T.; Wang, B.; Yancopoulou, D.; Ricklin, D.; Lambris, J.D. Complement inhibition in pre-clinical models of periodontitis and prospects for clinical application. Semin. Immunol. 2016, 28, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Reis, E.S.; DeAngelis, R.A.; Chen, H.; Resuello, R.R.; Ricklin, D.; Lambris, J.D. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology 2014, 220, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueras-Ortiz, C.J.; Mahairaki, V.; Delgado-Peraza, F.; Das, D.; Avgerinos, K.; Eren, E.; Hentschel, M.; Goetzl, E.J.; Mattson, M.P.; Kapogiannis, D. Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells 2020, 9, 1618. https://doi.org/10.3390/cells9071618
Nogueras-Ortiz CJ, Mahairaki V, Delgado-Peraza F, Das D, Avgerinos K, Eren E, Hentschel M, Goetzl EJ, Mattson MP, Kapogiannis D. Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells. 2020; 9(7):1618. https://doi.org/10.3390/cells9071618
Chicago/Turabian StyleNogueras-Ortiz, Carlos J., Vasiliki Mahairaki, Francheska Delgado-Peraza, Debamitra Das, Konstantinos Avgerinos, Erden Eren, Matthew Hentschel, Edward J. Goetzl, Mark P. Mattson, and Dimitrios Kapogiannis. 2020. "Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity" Cells 9, no. 7: 1618. https://doi.org/10.3390/cells9071618
APA StyleNogueras-Ortiz, C. J., Mahairaki, V., Delgado-Peraza, F., Das, D., Avgerinos, K., Eren, E., Hentschel, M., Goetzl, E. J., Mattson, M. P., & Kapogiannis, D. (2020). Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells, 9(7), 1618. https://doi.org/10.3390/cells9071618