Revealing the Proteome of Motor Cortex Derived Extracellular Vesicles Isolated from Amyotrophic Lateral Sclerosis Human Postmortem Tissues

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MCEV Isolation from Brain Tissue
2.2. SDS-PAGE Gel Electrophoresis
2.3. Nanoparticle Tracking Analysis
2.4. Transmission Electron Microscopy
2.5. Mass Spectrometry
2.6. Data Analysis and Protein Identification
3. Results
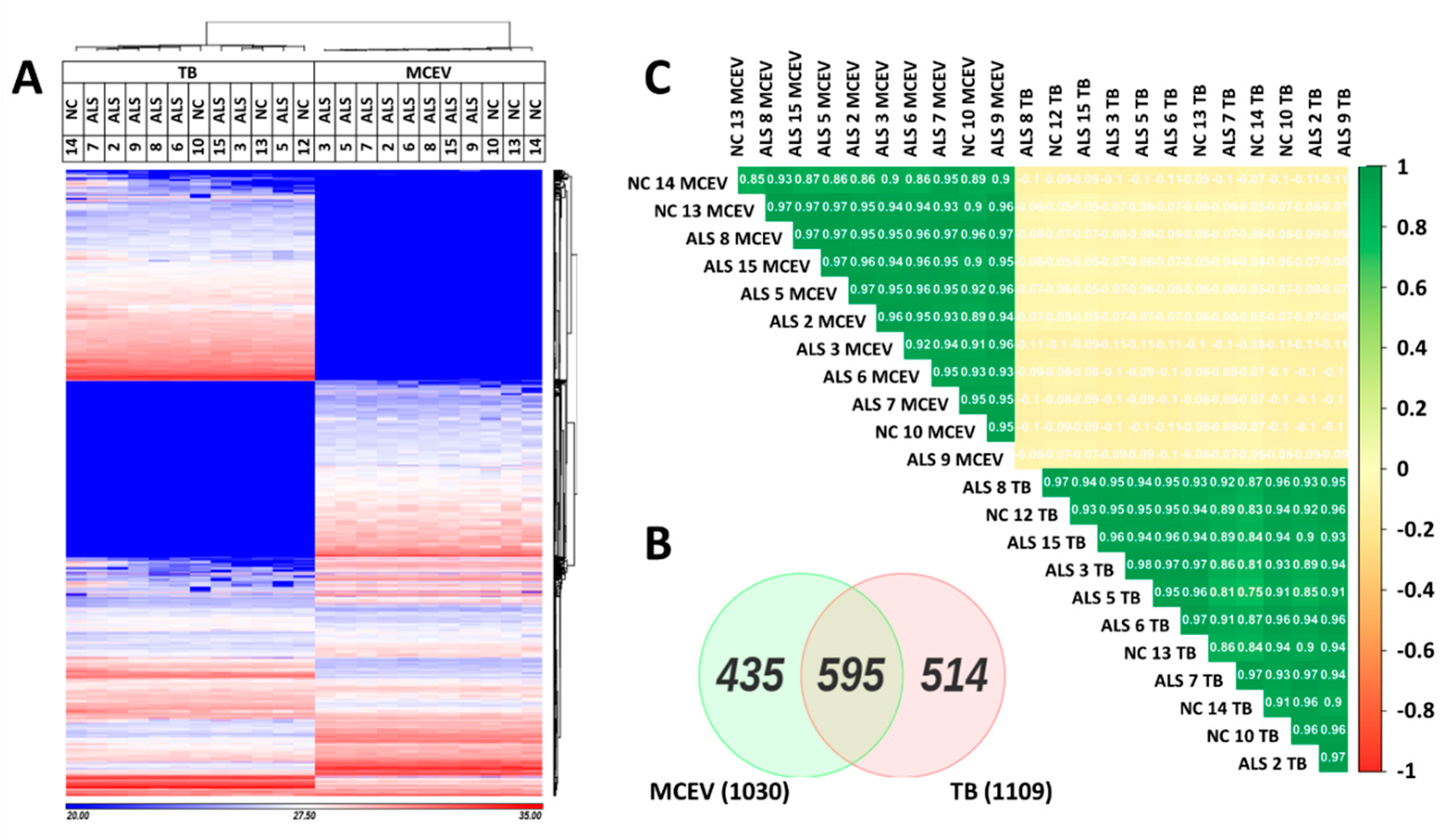
3.1. MCEVs Isolated Display the Characteristics of Small EVs
3.2. MCEVs Display a Unique Proteome Compared to Their respective TBs
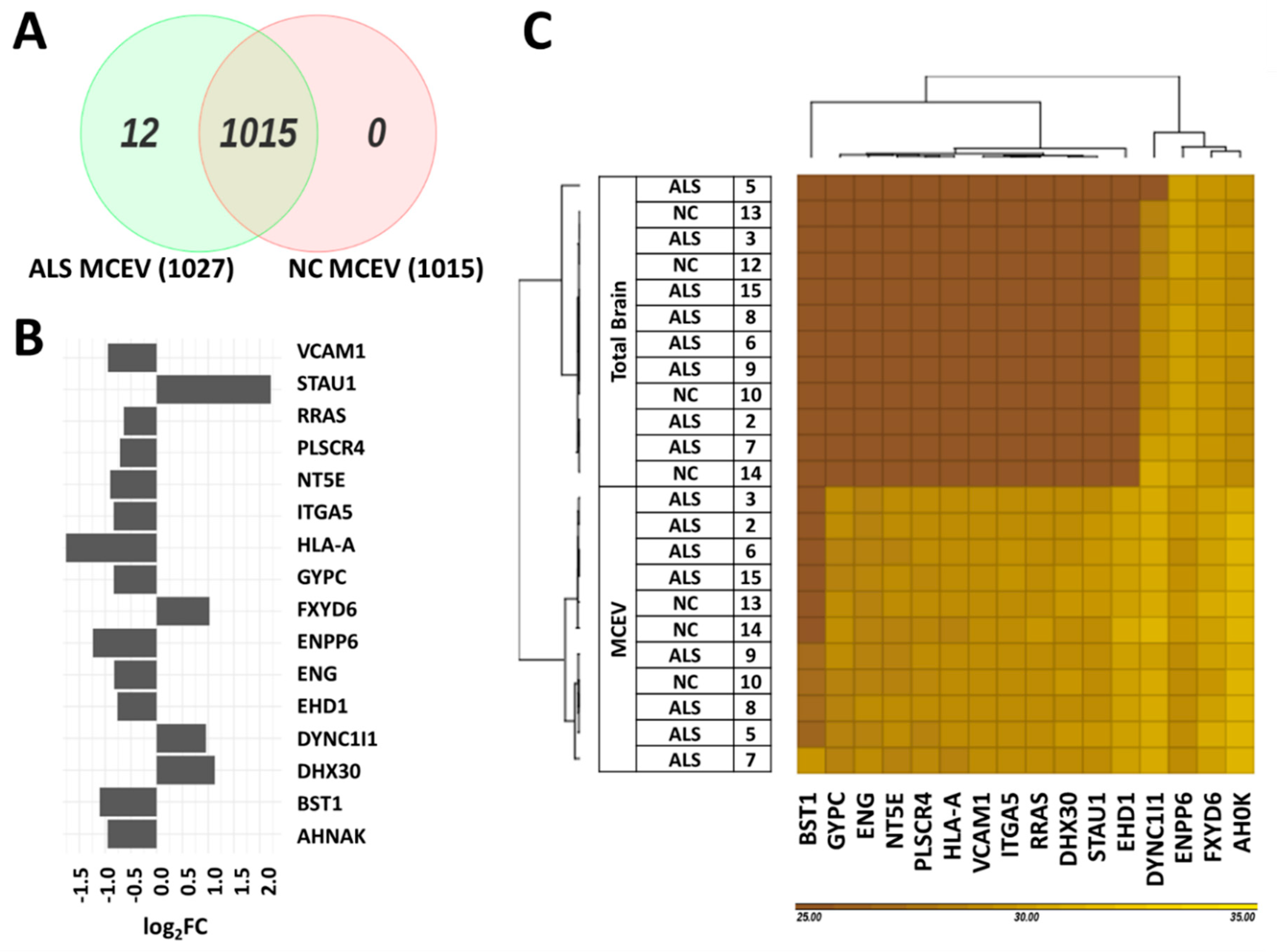
3.3. Differentially Expressed Proteins Found in ALS MCEVs Compared to NC MCEVs
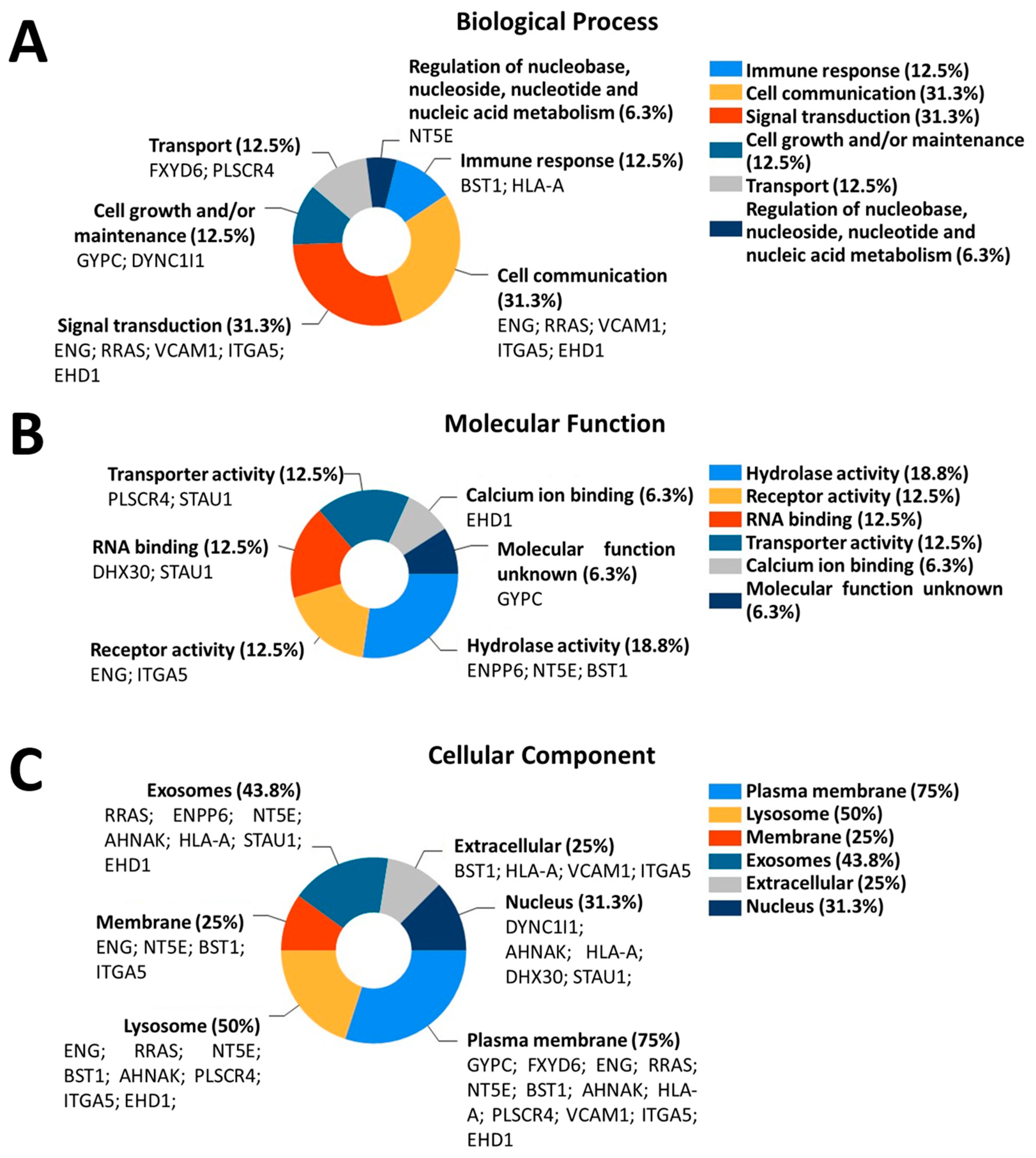
3.4. Differentially Expressed Proteins in ALS MCEVs Are Involved in Communication, Exhibit RNA-Binding Activity and Are Sequestered to Lysosomes and Exosomes
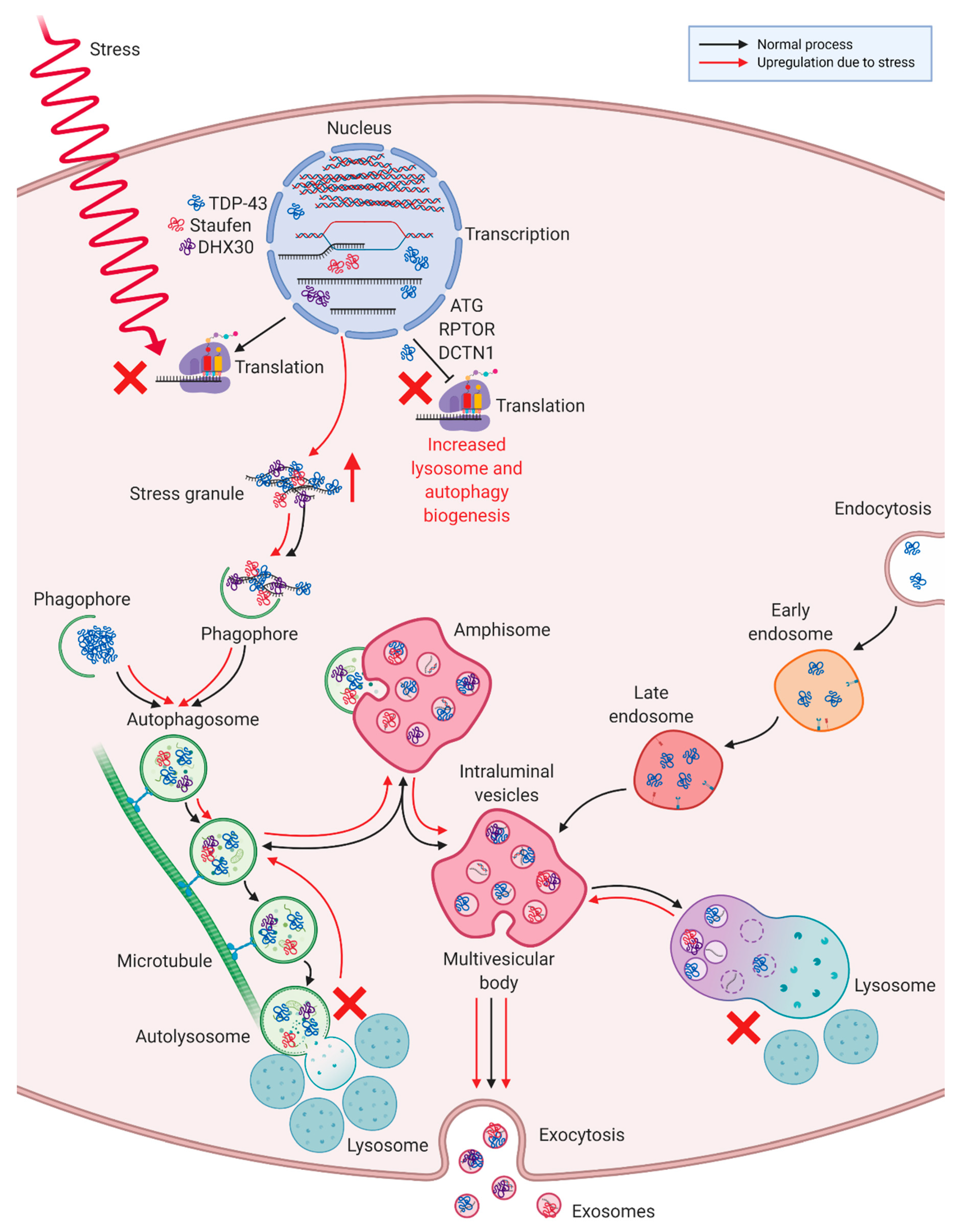
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic Proteins in Neurodegenerative Disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.M.; Che, C.H.; Feng, S.Y.; Liu, C.Y.; Li, L.Y.; Li, Y.X.; Huang, H.P.; Zou, Z.Y. Novel mutation in optineurin causing aggressive ALS+/-frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2377–2383. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wu, W.; Xie, Z.; Gang, Q.; Yu, M.; Liu, J.; Wang, Q.; Lv, H.; Zhang, W.; Huang, Y.; et al. Novel and Recurrent Mutations in a Cohort of Chinese Patients With Young-Onset Amyotrophic Lateral Sclerosis. Front. Neurosci. 2019, 13, 1289. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Sprovieri, T.; Ungaro, C.; Perrone, B.; Naimo, G.D.; Spataro, R.; Cavallaro, S.; La Bella, V.; Conforti, F.L. A novel S379A TARDBP mutation associated to late-onset sporadic ALS. Neurol. Sci. 2019, 40, 2111–2118. [Google Scholar] [CrossRef]
- Corcia, P.; Danel, V.; Lacour, A.; Beltran, S.; Andres, C.; Couratier, P.; Blasco, H.; Vourc’h, P. A novel mutation of the C-terminal amino acid of FUS (Y526C) strengthens FUS gene as the most frequent genetic factor in aggressive juvenile ALS. Amyotroph Lateral Scler Front. Degener 2017, 18, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Ayala, V.; Granado-Serrano, A.B.; Cacabelos, D.; Naudi, A.; Ilieva, E.V.; Boada, J.; Caraballo-Miralles, V.; Llado, J.; Ferrer, I.; Pamplona, R.; et al. Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: Implications in ALS. Acta Neuropathol. 2011, 122, 259–270. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Figueroa, K.P.; Scoles, D.R.; Pulst, S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 2018, 9, 3648. [Google Scholar] [CrossRef] [PubMed]
- Kiebler, M.A.; Hemraj, I.; Verkade, P.; Kohrmann, M.; Fortes, P.; Marion, R.M.; Ortin, J.; Dotti, C.G. The mammalian staufen protein localizes to the somatodendritic domain of cultured hippocampal neurons: Implications for its involvement in mRNA transport. J. Neurosci. 1999, 19, 288–297. [Google Scholar] [CrossRef]
- Lessel, D.; Schob, C.; Küry, S.; Reijnders, M.R.F.; Harel, T.; Eldomery, M.K.; Coban-Akdemir, Z.; Denecke, J.; Edvardson, S.; Colin, E.; et al. De Novo Missense Mutations in DHX30 Impair Global Translation and Cause a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 101, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Lo Bello, M.; Di Fini, F.; Notaro, A.; Spataro, R.; Conforti, F.L.; La Bella, V. ALS-Related Mutant FUS Protein Is Mislocalized to Cytoplasm and Is Recruited into Stress Granules of Fibroblasts from Asymptomatic FUS P525L Mutation Carriers. Neuro-Degener. Dis. 2017, 17, 292–303. [Google Scholar] [CrossRef]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 Is Directed to Stress Granules by Sorbitol, a Novel Physiological Osmotic and Oxidative Stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J., Jr.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H., Jr.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [Green Version]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.; Chian, R.J.; Boggio, K.J.; Quaresma, A.J.; Nickerson, J.A.; Bosco, D.A. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [Green Version]
- Sama, R.R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Li, C.; Zhang, Y.; Zhang, D.; Otterbein, L.E.; Jin, Y. Caveolin-1 selectively regulates microRNA sorting into microvesicles after noxious stimuli. J. Exp. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Abramowicz, A.; Widłak, P.; Pietrowska, M. Different Types of Cellular Stress Affect the Proteome Composition of Small Extracellular Vesicles: A Mini Review. Proteomes 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [Green Version]
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurrò, M.R.; et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 2019, 9, e028486. [Google Scholar] [CrossRef]
- De Jong, O.G.; Verhaar, M.C.; Chen, Y.; Vader, P.; Gremmels, H.; Posthuma, G.; Schiffelers, R.M.; Gucek, M.; van Balkom, B.W.M. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collen, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef] [Green Version]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borras, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoorvogel, W.; Strous, G.J.; Geuze, H.J.; Oorschot, V.; Schwartz, A.L. Late endosomes derive from early endosomes by maturation. Cell 1991, 65, 417–427. [Google Scholar] [CrossRef]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural. Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Buschow, S.I.; Nolte-’t Hoen, E.N.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Vella, L.J.; Sharples, R.A.; Lawson, V.A.; Masters, C.L.; Cappai, R.; Hill, A.F. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 2007, 211, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Scicluna, B.J.; Cheng, L.; Bawden, E.G.; Masters, C.L.; Ang, C.-S.; Willamson, N.; McLean, C.; Barnham, K.J.; Hill, A.F. A rigorous method to enrich for exosomes from brain tissue. J. Extracell. Vesicles 2017, 6, 1348885. [Google Scholar] [CrossRef] [PubMed]
- Müller-Taubenberger, A.; Lupas, A.N.; Li, H.; Ecke, M.; Simmeth, E.; Gerisch, G. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 2001, 20, 6772–6782. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Deng, W.; Klinke, D.J. Exosomes: Improved methods to characterize their morphology, RNA content, and surface protein biomarkers. Analyst 2015, 140, 6631–6642. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef] [Green Version]
- Pathan, M.; Keerthikumar, S.; Ang, C.-S.; Gangoda, L.; Quek, C.Y.J.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [Green Version]
- Coleman, B.M.; Hanssen, E.; Lawson, V.A.; Hill, A.F. Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. FASEB J. 2012, 26, 4160–4173. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Soc. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [Green Version]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Hamlett, E.D.; Ledreux, A.; Potter, H.; Chial, H.J.; Patterson, D.; Espinosa, J.M.; Bettcher, B.M.; Granholm, A.C. Exosomal biomarkers in Down syndrome and Alzheimer’s disease. Free Radic. Biol. Med. 2018, 114, 110–121. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.F.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef] [Green Version]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Et Biophys. Acta 2016, 1862, 2004–2014. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, K.; Chen, L.; Fan, D. Increased Interleukin-6 Levels in the Astrocyte-Derived Exosomes of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182. [Google Scholar] [CrossRef]
- Moisse, K.; Volkening, K.; Leystra-Lantz, C.; Welch, I.; Hill, T.; Strong, M.J. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: Implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009, 1249, 202–211. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef] [Green Version]
- Gershoni-Emek, N.; Mazza, A.; Chein, M.; Gradus-Pery, T.; Xiang, X.; Li, K.W.; Sharan, R.; Perlson, E. Proteomic Analysis of Dynein-Interacting Proteins in Amyotrophic Lateral Sclerosis Synaptosomes Reveals Alterations in the RNA-Binding Protein Staufen1. Mol. Cell Proteom. 2016, 15, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.R.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph Lateral Scler Front. Degener 2014, 15, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Haraldsen, G.; Kvale, D.; Lien, B.; Farstad, I.N.; Brandtzaeg, P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J. Immunol 1996, 156, 2558–2565. [Google Scholar] [PubMed]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.C.; Serres, S.; Khrapitchev, A.A.; Stolp, H.B.; Anthony, D.C.; Talbot, K.; Turner, M.R.; Sibson, N.R. T₂-weighted MRI detects presymptomatic pathology in the SOD1 mouse model of ALS. J. Cereb. Blood Flow Metab. 2014, 34, 785–793. [Google Scholar] [CrossRef]
- Kunis, G.; Baruch, K.; Miller, O.; Schwartz, M. Immunization with a Myelin-Derived Antigen Activates the Brain’s Choroid Plexus for Recruitment of Immunoregulatory Cells to the CNS and Attenuates Disease Progression in a Mouse Model of ALS. J. Neurosci. 2015, 35, 6381–6393. [Google Scholar] [CrossRef] [PubMed]
- Iłzecka, J. Decreased serum endoglin level in patients with amyotrophic lateral sclerosis: A preliminary report. Scand J. Clin. Lab. Investig. 2008, 68, 348–351. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Ehrhart, J.; Mustafa, H.; Llauget, A.; Boccio, K.J.; Sanberg, P.R.; Appel, S.H.; Borlongan, C.V. Phenotypic characteristics of human bone marrow-derived endothelial progenitor cells in vitro support cell effectiveness for repair of the blood-spinal cord barrier in ALS. Brain Res. 2019, 1724, 146428. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, X.; Liu, Y.; Gong, C.; Wei, X.; Zhang, T.; Ma, D.; Gao, Q. Activation of integrin beta1 mediates the increased malignant potential of ovarian cancer cells exerted by inflammatory cytokines. Anticancer Agents Med. Chem. 2014, 14, 955–962. [Google Scholar] [CrossRef]
- Lehto, M.; Mäyränpää, M.I.; Pellinen, T.; Ihalmo, P.; Lehtonen, S.; Kovanen, P.T.; Groop, P.-H.; Ivaska, J.; Olkkonen, V.M. The R-Ras interaction partner ORP3 regulates cell adhesion. J. Cell Sci. 2008, 121, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef]
- Volonté, C.; Apolloni, S.; Carrì, M.T.; D’Ambrosi, N. ALS: Focus on purinergic signalling. Pharmacol. Ther. 2011, 132, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Cieślak, M.; Roszek, K.; Wujak, M. Purinergic implication in amyotrophic lateral sclerosis—From pathological mechanisms to therapeutic perspectives. Purinergic Signal. 2019, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrowolny, G.; Bernardini, C.; Martini, M.; Baranzini, M.; Barba, M.; Musarò, A. Muscle Expression of SOD1(G93A) Modulates microRNA and mRNA Transcription Pattern Associated with the Myelination Process in the Spinal Cord of Transgenic Mice. Front. Cell. Neurosci. 2015, 9, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef]
- Štalekar, M.; Yin, X.; Rebolj, K.; Darovic, S.; Troakes, C.; Mayr, M.; Shaw, C.E.; Rogelj, B. Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 2015, 293, 157–170. [Google Scholar] [CrossRef]
- Hedlund, E.; Karlsson, M.; Osborn, T.; Ludwig, W.; Isacson, O. Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 2010, 133, 2313–2330. [Google Scholar] [CrossRef] [Green Version]
- Andrés-Benito, P.; Moreno, J.; Aso, E.; Povedano, M.; Ferrer, I. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: Implications in frontotemporal lobar degeneration. Aging 2017, 9, 823–851. [Google Scholar] [CrossRef] [Green Version]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerod, L.; Fisher, E.M.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. How do ESCRT proteins control autophagy? J. Cell Sci. 2009, 122, 2179–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. CB 2007, 17, 1561–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrow, L.; Malhotra, R.; Debnath, J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 2015, 17, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Song, C.-Y.; Guo, J.-F.; Liu, Y.; Tang, B.-S. Autophagy and Its Comprehensive Impact on ALS. Int. J. Neurosci. 2012, 122, 695–703. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, J.H.; Lee, J.E.; Choi, E.-J. Role of autophagy in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Et Biophys. Acta 2015, 1852, 2517–2524. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016, 1649, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Bose, J.K.; Huang, C.-C.; Shen, C.K.J. Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.-P. Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Otake, K.; Kamiguchi, H.; Hirozane, Y. Identification of biomarkers for amyotrophic lateral sclerosis by comprehensive analysis of exosomal mRNAs in human cerebrospinal fluid. BMC Med. Genom. 2019, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassileff, N.; Vella, L.J.; Rajapaksha, H.; Shambrook, M.; Kenari, A.N.; McLean, C.; Hill, A.F.; Cheng, L. Revealing the Proteome of Motor Cortex Derived Extracellular Vesicles Isolated from Amyotrophic Lateral Sclerosis Human Postmortem Tissues. Cells 2020, 9, 1709. https://doi.org/10.3390/cells9071709
Vassileff N, Vella LJ, Rajapaksha H, Shambrook M, Kenari AN, McLean C, Hill AF, Cheng L. Revealing the Proteome of Motor Cortex Derived Extracellular Vesicles Isolated from Amyotrophic Lateral Sclerosis Human Postmortem Tissues. Cells. 2020; 9(7):1709. https://doi.org/10.3390/cells9071709
Chicago/Turabian StyleVassileff, Natasha, Laura J. Vella, Harinda Rajapaksha, Mitch Shambrook, Amirmohammad Nasiri Kenari, Catriona McLean, Andrew F. Hill, and Lesley Cheng. 2020. "Revealing the Proteome of Motor Cortex Derived Extracellular Vesicles Isolated from Amyotrophic Lateral Sclerosis Human Postmortem Tissues" Cells 9, no. 7: 1709. https://doi.org/10.3390/cells9071709
APA StyleVassileff, N., Vella, L. J., Rajapaksha, H., Shambrook, M., Kenari, A. N., McLean, C., Hill, A. F., & Cheng, L. (2020). Revealing the Proteome of Motor Cortex Derived Extracellular Vesicles Isolated from Amyotrophic Lateral Sclerosis Human Postmortem Tissues. Cells, 9(7), 1709. https://doi.org/10.3390/cells9071709