Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity





Abstract
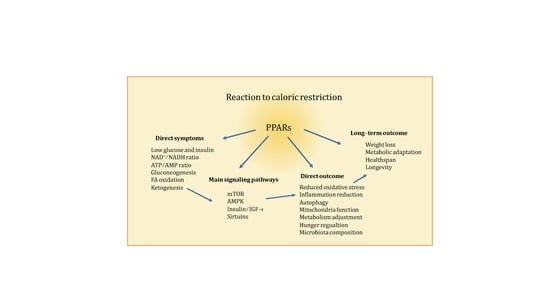
:
{kind=link}
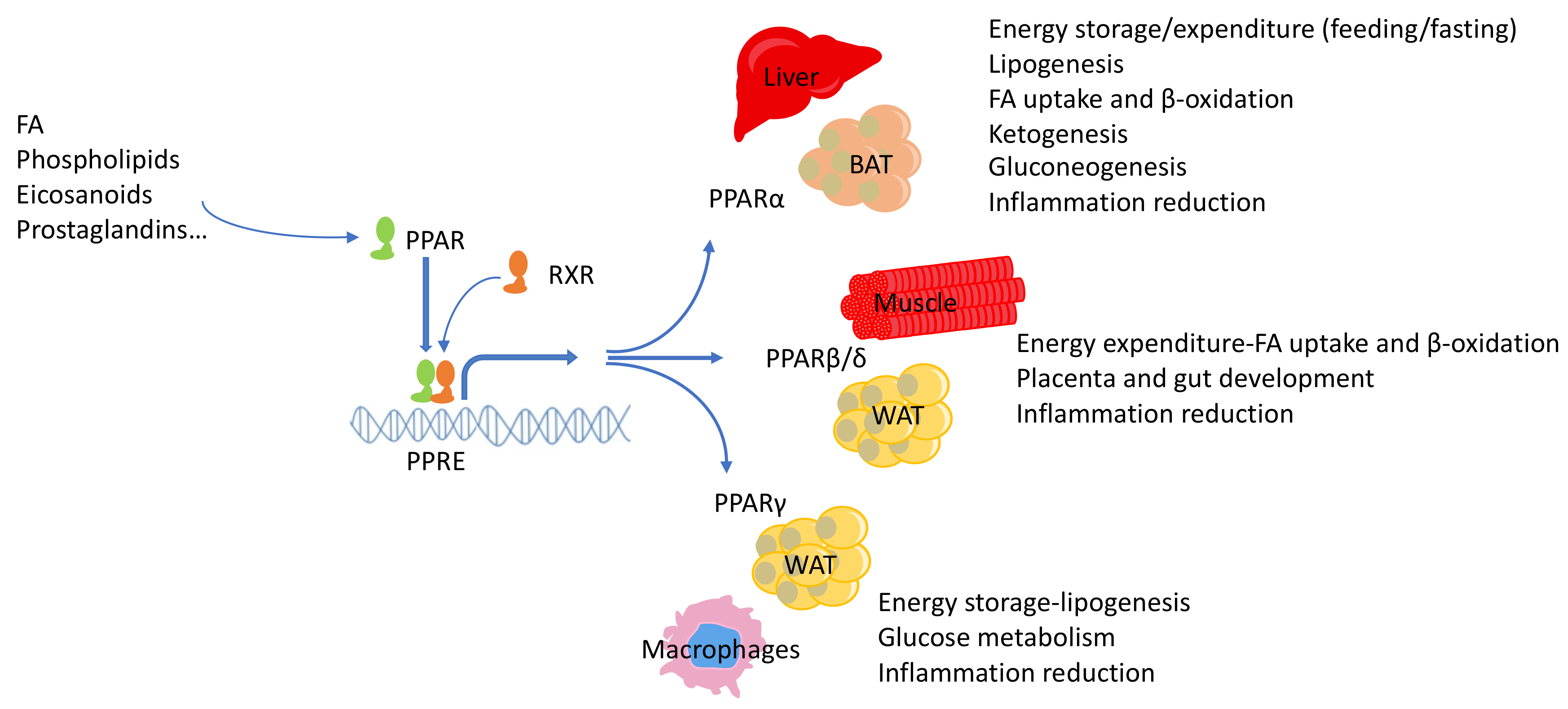{kind=link}
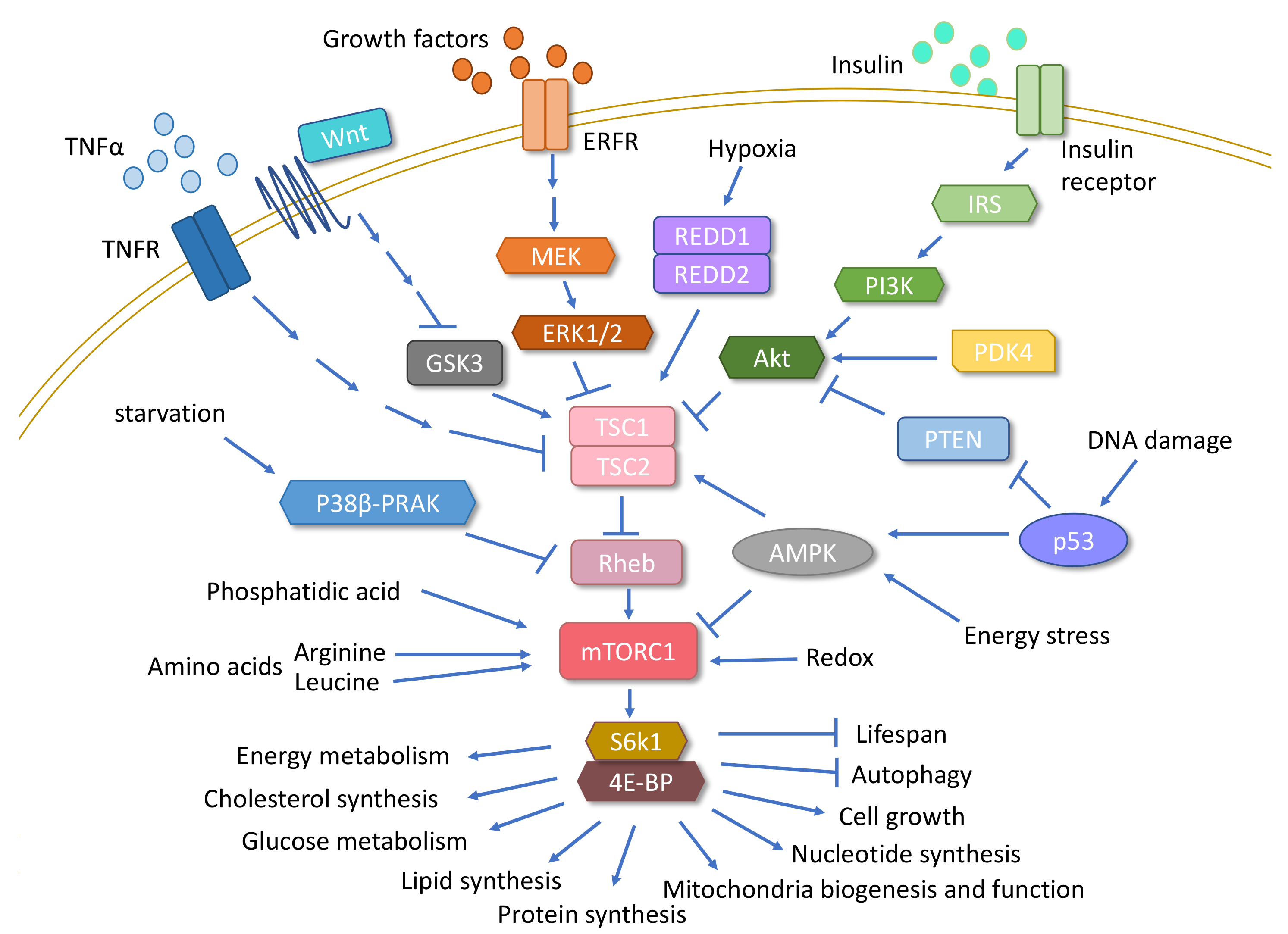{kind=link}
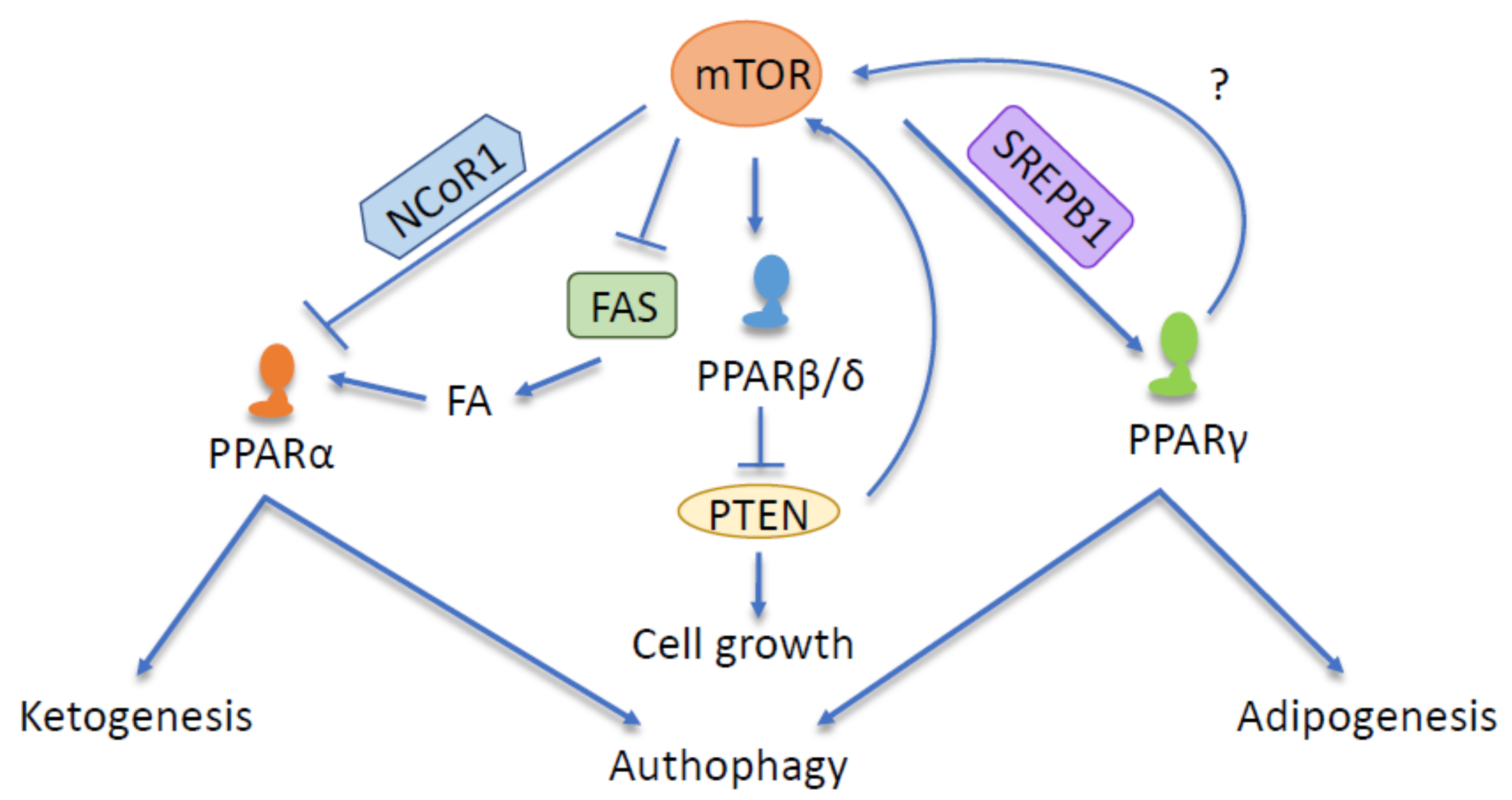{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. PPARs
2.1. PPARα
2.2. PPARβ/δ
2.3. PPARγ
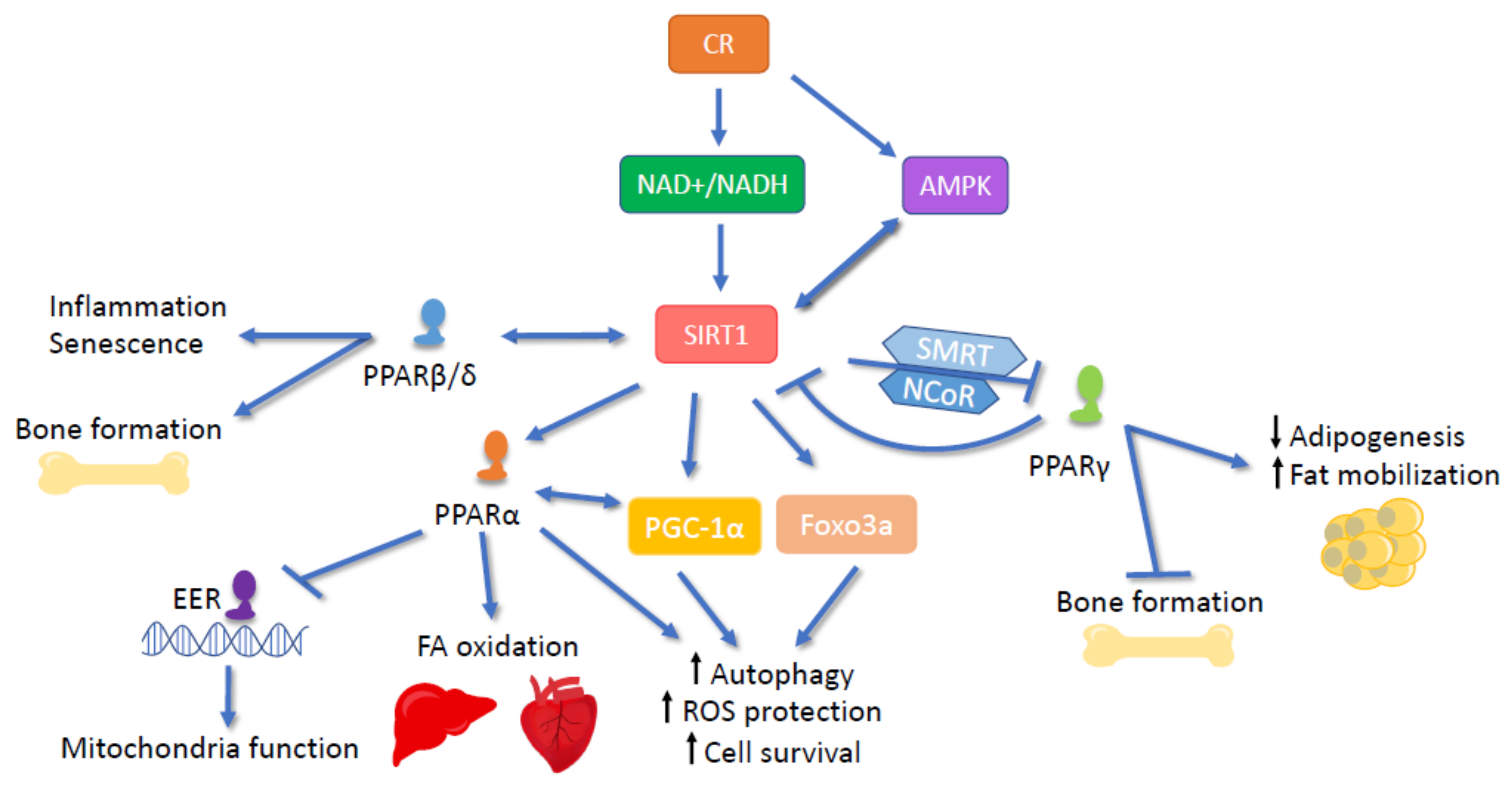
2.4. PPARs in CR
3. Major Pathways Affected by CR
3.1. mTOR
3.2. mTOR and PPARα
3.3. mTOR and PPARβ/δ
3.4. mTOR and PPARγ
4. AMPK
4.1. AMPK and PPARα
4.2. AMPK and PPARβ/δ
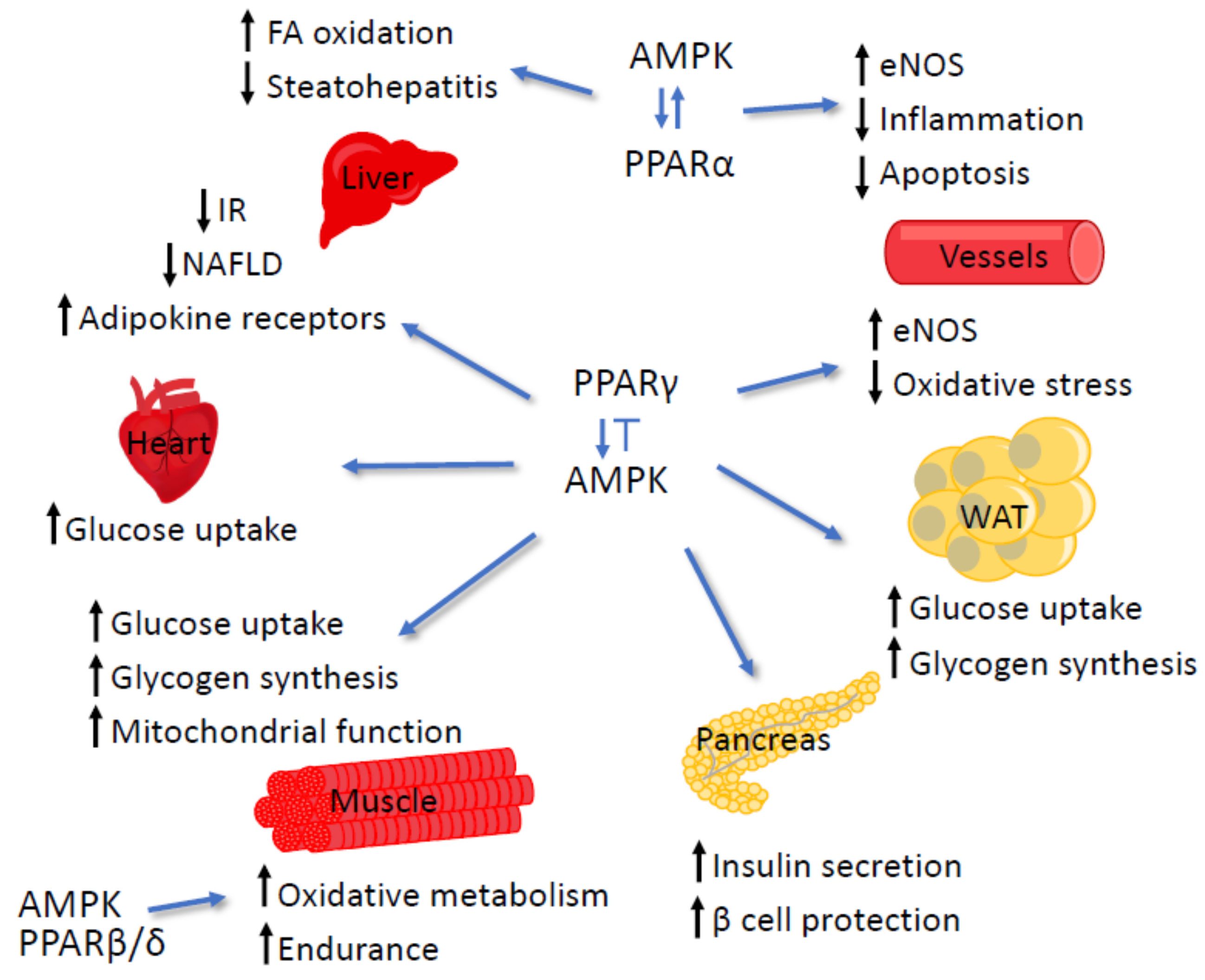
4.3. AMPK and PPARγ
5. Insulin Signaling
5.1. Insulin Signaling and PPARα
5.2. Insulin Signaling and PPARβ/δ
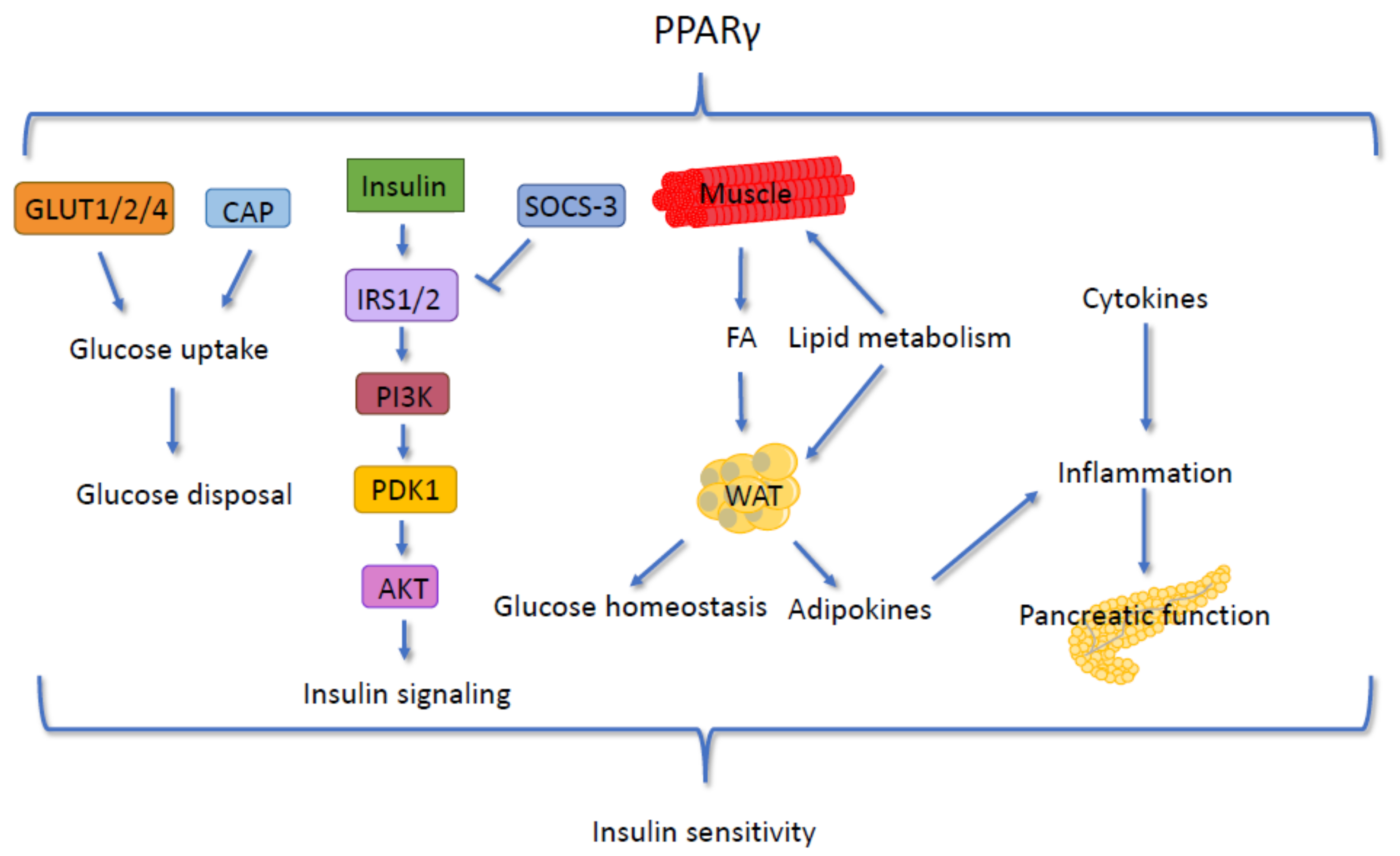
5.3. Insulin Signaling and PPARγ
6. Sirtuins
6.1. SIRT and PPARα
6.2. SIRT and PPARβ/δ
6.3. SIRT1 and PPARγ
7. Major Outcomes of CR
7.1. Oxidative Stress Reduction
7.2. Mitochondrial Function
7.3. Reduction of Inflammation
7.4. Metabolic Adaptation
7.5. Physical Exercise
7.6. Hunger
7.7. Longevity and Aging
7.8. Microbiota Composition
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Osborne, T.B.; Mendel, L.B.; Ferry, E.L. The Effect of Retardation of Growth Upon the Breeding Period and Duration of Life of Rats. Science 1917, 45, 294–295. [Google Scholar] [CrossRef] [Green Version]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 1989, 5, 155–171, discussion 172. [Google Scholar]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef]
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Asp. Med. 2011, 32, 159–221. [Google Scholar] [CrossRef]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.M.; Shanmuganayagam, D.; Weindruch, R. Caloric restriction and aging: Studies in mice and monkeys. Toxicol. Pathol. 2009, 37, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6659–6663. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Klein, S. Aging, adiposity, and calorie restriction. JAMA 2007, 297, 986–994. [Google Scholar] [CrossRef]
- Cerqueira, F.M.; Kowaltowski, A.J. Commonly adopted caloric restriction protocols often involve malnutrition. Ageing Res. Rev. 2010, 9, 424–430. [Google Scholar] [CrossRef]
- Dogan, S.; Ray, A.; Cleary, M.P. The influence of different calorie restriction protocols on serum pro-inflammatory cytokines, adipokines and IGF-I levels in female C57BL6 mice: Short term and long term diet effects. Meta Gene 2017, 12, 22–32. [Google Scholar] [CrossRef]
- Dogan, S.; Rogozina, O.P.; Lokshin, A.E.; Grande, J.P.; Cleary, M.P. Effects of chronic vs. intermittent calorie restriction on mammary tumor incidence and serum adiponectin and leptin levels in MMTV-TGF-alpha mice at different ages. Oncol. Lett. 2010, 1, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Phelan, J.P.; Rose, M.R. Why dietary restriction substantially increases longevity in animal models but won’t in humans. Ageing Res. Rev. 2005, 4, 339–350. [Google Scholar] [CrossRef]
- Chung, H.Y.; Kim, H.J.; Kim, J.W.; Yu, B.P. The inflammation hypothesis of aging: Molecular modulation by calorie restriction. Ann. N. Y. Acad. Sci. 2001, 928, 327–335. [Google Scholar]
- Nuclear Receptors Nomenclature, C. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-Regulated Gene Expression. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Horlein, A.J.; Naar, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef]
- Chen, J.D.; Evans, R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar] [CrossRef]
- Kang, Z.; Fan, R. PPARalpha and NCOR/SMRT corepressor network in liver metabolic regulation. FASEB J. 2020. [Google Scholar] [CrossRef]
- Yu, C.; Markan, K.; Temple, K.A.; Deplewski, D.; Brady, M.J.; Cohen, R.N. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J. Biol. Chem. 2005, 280, 13600–13605. [Google Scholar] [CrossRef] [Green Version]
- Krogsdam, A.M.; Nielsen, C.A.; Neve, S.; Holst, D.; Helledie, T.; Thomsen, B.; Bendixen, C.; Mandrup, S.; Kristiansen, K. Nuclear receptor corepressor-dependent repression of peroxisome-proliferator-activated receptor delta-mediated transactivation. Biochem. J. 2002, 363, 157–165. [Google Scholar] [CrossRef]
- Neschen, S.; Morino, K.; Dong, J.; Wang-Fischer, Y.; Cline, G.W.; Romanelli, A.J.; Rossbacher, J.C.; Moore, I.K.; Regittnig, W.; Munoz, D.S.; et al. n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes 2007, 56, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef]
- Plutzky, J. Peroxisome proliferator-activated receptors in vascular biology and atherosclerosis: Emerging insights for evolving paradigms. Curr. Atheroscler. Rep. 2000, 2, 327–335. [Google Scholar]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Moller, D.E.; Berger, J.P. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int. J. Obes. Relat. Metab. Disord. 2003, 27 (Suppl. S3), S17–S21. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural Peroxisome Proliferator-Activated Receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARalpha/gamma agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [Green Version]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) alpha/gamma/delta Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [Green Version]
- Green, S.; Wahli, W. Peroxisome proliferator-activated receptors: Finding the orphan a home. Mol. Cell Endocrinol. 1994, 100, 149–153. [Google Scholar]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Lazennec, G.; Canaple, L.; Saugy, D.; Wahli, W. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol. Endocrinol. 2000, 14, 1962–1975. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Willis, M.S. The story so far: Post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H515–H526. [Google Scholar] [CrossRef] [Green Version]
- Floyd, Z.E.; Stephens, J.M. Controlling a master switch of adipocyte development and insulin sensitivity: Covalent modifications of PPARgamma. Biochim. Biophys. Acta 2012, 1822, 1090–1095. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef] [Green Version]
- Diradourian, C.; Girard, J.; Pegorier, J.P. Phosphorylation of PPARs: From molecular characterization to physiological relevance. Biochimie 2005, 87, 33–38. [Google Scholar] [CrossRef]
- Leuenberger, N.; Pradervand, S.; Wahli, W. Sumoylated PPARalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J. Clin. Investig. 2009, 119, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Robitaille, J.; Brouillette, C.; Houde, A.; Lemieux, S.; Perusse, L.; Tchernof, A.; Gaudet, D.; Vohl, M.C. Association between the PPARalpha-L162V polymorphism and components of the metabolic syndrome. J. Hum. Genet. 2004, 49, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Lacquemant, C.; Lepretre, F.; Pineda Torra, I.; Manraj, M.; Charpentier, G.; Ruiz, J.; Staels, B.; Froguel, P. Mutation screening of the PPARalpha gene in type 2 diabetes associated with coronary heart disease. Diabetes Metab. 2000, 26, 393–401. [Google Scholar]
- Vohl, M.C.; Lepage, P.; Gaudet, D.; Brewer, C.G.; Betard, C.; Perron, P.; Houde, G.; Cellier, C.; Faith, J.M.; Despres, J.P.; et al. Molecular scanning of the human PPARa gene: Association of the L162v mutation with hyperapobetalipoproteinemia. J. Lipid Res. 2000, 41, 945–952. [Google Scholar]
- Flavell, D.M.; Pineda Torra, I.; Jamshidi, Y.; Evans, D.; Diamond, J.R.; Elkeles, R.S.; Bujac, S.R.; Miller, G.; Talmud, P.J.; Staels, B.; et al. Variation in the PPARalpha gene is associated with altered function in vitro and plasma lipid concentrations in Type II diabetic subjects. Diabetologia 2000, 43, 673–680. [Google Scholar] [CrossRef]
- Tanaka, T.; Ordovas, J.M.; Delgado-Lista, J.; Perez-Jimenez, F.; Marin, C.; Perez-Martinez, P.; Gomez, P.; Lopez-Miranda, J. Peroxisome proliferator-activated receptor alpha polymorphisms and postprandial lipemia in healthy men. J. Lipid Res. 2007, 48, 1402–1408. [Google Scholar] [CrossRef] [Green Version]
- Andrulionyte, L.; Kuulasmaa, T.; Chiasson, J.L.; Laakso, M.; Group, S.-N.S. Single nucleotide polymorphisms of the peroxisome proliferator-activated receptor-alpha gene (PPARA) influence the conversion from impaired glucose tolerance to type 2 diabetes: The STOP-NIDDM trial. Diabetes 2007, 56, 1181–1186. [Google Scholar] [CrossRef] [Green Version]
- Flavell, D.M.; Ireland, H.; Stephens, J.W.; Hawe, E.; Acharya, J.; Mather, H.; Hurel, S.J.; Humphries, S.E. Peroxisome proliferator-activated receptor alpha gene variation influences age of onset and progression of type 2 diabetes. Diabetes 2005, 54, 582–586. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: The PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef] [Green Version]
- Smati, S.; Regnier, M.; Fougeray, T.; Polizzi, A.; Fougerat, A.; Lasserre, F.; Lukowicz, C.; Tramunt, B.; Guillaume, M.; Burnol, A.F.; et al. Regulation of hepatokine gene expression in response to fasting and feeding: Influence of PPAR-alpha and insulin-dependent signalling in hepatocytes. Diabetes Metab. 2019. [Google Scholar] [CrossRef]
- Paumelle, R.; Haas, J.T.; Hennuyer, N.; Bauge, E.; Deleye, Y.; Mesotten, D.; Langouche, L.; Vanhoutte, J.; Cudejko, C.; Wouters, K.; et al. Hepatic PPARalpha is critical in the metabolic adaptation to sepsis. J. Hepatol. 2019, 70, 963–973. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.M.; Hu, M.; Chan, P.; Tomlinson, B. Early investigational drugs targeting PPAR-alpha for the treatment of metabolic disease. Expert Opin. Investig. Drugs 2015, 24, 611–621. [Google Scholar] [CrossRef]
- Taniguchi, A.; Fukushima, M.; Sakai, M.; Tokuyama, K.; Nagata, I.; Fukunaga, A.; Kishimoto, H.; Doi, K.; Yamashita, Y.; Matsuura, T.; et al. Effects of bezafibrate on insulin sensitivity and insulin secretion in non-obese Japanese type 2 diabetic patients. Metabolism 2001, 50, 477–480. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Staels, B.; Duriez, P. The role of fibric acids in atherosclerosis. Curr. Atheroscler. Rep. 2001, 3, 83–92. [Google Scholar]
- Fruchart, J.C.; Santos, R.D.; Aguilar-Salinas, C.; Aikawa, M.; Al Rasadi, K.; Amarenco, P.; Barter, P.J.; Ceska, R.; Corsini, A.; Despres, J.P.; et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha) paradigm: Conceptual framework and therapeutic potential: A consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc. Diabetol. 2019, 18, 71. [Google Scholar] [CrossRef]
- Girroir, E.E.; Hollingshead, H.E.; He, P.; Zhu, B.; Perdew, G.H.; Peters, J.M. Quantitative expression patterns of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) protein in mice. Biochem. Biophys. Res. Commun. 2008, 371, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Braissant, O.; Wahli, W. Differential expression of peroxisome proliferator-activated receptor-alpha, -beta, and -gamma during rat embryonic development. Endocrinology 1998, 139, 2748–2754. [Google Scholar] [CrossRef]
- Tan, N.S.; Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor (PPAR)-beta as a target for wound healing drugs: What is possible? Am. J. Clin. Dermatol. 2003, 4, 523–530. [Google Scholar] [CrossRef]
- Di-Poi, N.; Tan, N.S.; Michalik, L.; Wahli, W.; Desvergne, B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol. Cell 2002, 10, 721–733. [Google Scholar] [CrossRef]
- Letavernier, E.; Perez, J.; Joye, E.; Bellocq, A.; Fouqueray, B.; Haymann, J.P.; Heudes, D.; Wahli, W.; Desvergne, B.; Baud, L. Peroxisome proliferator-activated receptor beta/delta exerts a strong protection from ischemic acute renal failure. J. Am. Soc. Nephrol. 2005, 16, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Michalik, L.; Wahli, W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J. Clin. Investig. 2006, 116, 598–606. [Google Scholar] [CrossRef]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol. Cell Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef] [Green Version]
- Barak, Y.; Liao, D.; He, W.; Ong, E.S.; Nelson, M.C.; Olefsky, J.M.; Boland, R.; Evans, R.M. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Nadra, K.; Anghel, S.I.; Joye, E.; Tan, N.S.; Basu-Modak, S.; Trono, D.; Wahli, W.; Desvergne, B. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor beta/delta. Mol. Cell Biol. 2006, 26, 3266–3281. [Google Scholar] [CrossRef] [Green Version]
- Varnat, F.; Heggeler, B.B.; Grisel, P.; Boucard, N.; Corthesy-Theulaz, I.; Wahli, W.; Desvergne, B. PPARbeta/delta regulates paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology 2006, 131, 538–553. [Google Scholar] [CrossRef]
- Doktorova, M.; Zwarts, I.; Zutphen, T.V.; Dijk, T.H.; Bloks, V.W.; Harkema, L.; Bruin, A.; Downes, M.; Evans, R.M.; Verkade, H.J.; et al. Intestinal PPARdelta protects against diet-induced obesity, insulin resistance and dyslipidemia. Sci. Rep. 2017, 7, 846. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Daoudi, M.; Hennuyer, N.; Borland, M.G.; Touche, V.; Duhem, C.; Gross, B.; Caiazzo, R.; Kerr-Conte, J.; Pattou, F.; Peters, J.M.; et al. PPARbeta/delta activation induces enteroendocrine L cell GLP-1 production. Gastroenterology 2011, 140, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor beta/delta in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [Green Version]
- Giordano Attianese, G.M.; Desvergne, B. Integrative and systemic approaches for evaluating PPARbeta/delta (PPARD) function. Nucl. Recept. Signal. 2015, 13, e001. [Google Scholar] [CrossRef] [Green Version]
- Anghel, S.I.; Wahli, W. Fat poetry: A kingdom for PPAR gamma. Cell Res. 2007, 17, 486–511. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Takakuwa, R.; Marchand, S.; Dentz, E.; Bornert, J.M.; Messaddeq, N.; Wendling, O.; Mark, M.; Desvergne, B.; Wahli, W.; et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 4543–4547. [Google Scholar] [CrossRef] [Green Version]
- Cerbone, A.; Toaldo, C.; Laurora, S.; Briatore, F.; Pizzimenti, S.; Dianzani, M.U.; Ferretti, C.; Barrera, G. 4-Hydroxynonenal and PPARgamma ligands affect proliferation, differentiation, and apoptosis in colon cancer cells. Free Radic. Biol. Med. 2007, 42, 1661–1670. [Google Scholar] [CrossRef]
- Martinasso, G.; Oraldi, M.; Trombetta, A.; Maggiora, M.; Bertetto, O.; Canuto, R.A.; Muzio, G. Involvement of PPARs in Cell Proliferation and Apoptosis in Human Colon Cancer Specimens and in Normal and Cancer Cell Lines. PPAR Res. 2007, 2007, 93416. [Google Scholar] [CrossRef] [Green Version]
- Theocharis, S.; Margeli, A.; Vielh, P.; Kouraklis, G. Peroxisome proliferator-activated receptor-gamma ligands as cell-cycle modulators. Cancer Treat. Rev. 2004, 30, 545–554. [Google Scholar] [CrossRef]
- Xu, W.P.; Zhang, X.; Xie, W.F. Differentiation therapy for solid tumors. J. Dig. Dis. 2014, 15, 159–165. [Google Scholar] [CrossRef]
- Chen, G.G.; Lee, J.F.; Wang, S.H.; Chan, U.P.; Ip, P.C.; Lau, W.Y. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and NF-kappaB in human colon cancer. Life Sci. 2002, 70, 2631–2646. [Google Scholar]
- Chen, G.G.; Xu, H.; Lee, J.F.; Subramaniam, M.; Leung, K.L.; Wang, S.H.; Chan, U.P.; Spelsberg, T.C. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor gamma-dependent pathway. Int. J. Cancer 2003, 107, 837–843. [Google Scholar] [CrossRef]
- Lee, C.J.; Han, J.S.; Seo, C.Y.; Park, T.H.; Kwon, H.C.; Jeong, J.S.; Kim, I.H.; Yun, J.; Bae, Y.S.; Kwak, J.Y.; et al. Pioglitazone, a synthetic ligand for PPARgamma, induces apoptosis in RB-deficient human colorectal cancer cells. Apoptosis Int. J. Program. Cell Death 2006, 11, 401–411. [Google Scholar] [CrossRef]
- Clay, C.E.; Monjazeb, A.; Thorburn, J.; Chilton, F.H.; High, K.P. 15-Deoxy-delta12,14-prostaglandin J2-induced apoptosis does not require PPARgamma in breast cancer cells. J. Lipid Res. 2002, 43, 1818–1828. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.F.; Zhang, Y.H.; Breyer, R.M.; Davis, L.; Breyer, M.D. Expression of peroxisome proliferator-activated receptor gamma (PPARgamma) in human transitional bladder cancer and its role in inducing cell death. Neoplasia 1999, 1, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J. Nuclear receptors. I. PPAR gamma in the gastrointestinal tract: Gain or pain? Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G581–G585. [Google Scholar] [CrossRef] [Green Version]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPARgamma and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Picard, F.; Auwerx, J. PPAR(gamma) and glucose homeostasis. Annu. Rev. Nutr. 2002, 22, 167–197. [Google Scholar] [CrossRef]
- Duszka, K.; Oresic, M.; Le May, C.; Konig, J.; Wahli, W. PPARgamma Modulates Long Chain Fatty Acid Processing in the Intestinal Epithelium. Int. J. Mol. Sci. 2017, 18, 2559. [Google Scholar] [CrossRef] [Green Version]
- Duszka, K.; Picard, A.; Ellero-Simatos, S.; Chen, J.; Defernez, M.; Paramalingam, E.; Pigram, A.; Vanoaica, L.; Canlet, C.; Parini, P.; et al. Intestinal PPARgamma signalling is required for sympathetic nervous system activation in response to caloric restriction. Sci. Rep. 2016, 6, 36937. [Google Scholar] [CrossRef]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.B.; Ducroc, R.; Regnault, B.; Kun Tan, C.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Watters, A.; Cheng, N.; Perry, C.E.; Xu, K.; Alicea, G.M.; Parris, J.L.D.; Baraban, E.; Ray, P.; Nayak, A.; et al. Polyunsaturated Fatty Acids from Astrocytes Activate PPARgamma Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 2019, 9, 1720–1735. [Google Scholar] [CrossRef] [Green Version]
- Patitucci, C.; Couchy, G.; Bagattin, A.; Caneque, T.; de Reynies, A.; Scoazec, J.Y.; Rodriguez, R.; Pontoglio, M.; Zucman-Rossi, J.; Pende, M.; et al. Hepatocyte nuclear factor 1alpha suppresses steatosis-associated liver cancer by inhibiting PPARgamma transcription. J. Clin. Investig. 2017, 127, 1873–1888. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Qi, C.; Korenberg, J.R.; Chen, X.N.; Noya, D.; Rao, M.S.; Reddy, J.K. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: Alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 7921–7925. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Elbrecht, A.; Chen, Y.; Cullinan, C.A.; Hayes, N.; Leibowitz, M.; Moller, D.E.; Berger, J. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem. Biophys. Res. Commun. 1996, 224, 431–437. [Google Scholar] [CrossRef]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar]
- Mukherjee, R.; Jow, L.; Croston, G.E.; Paterniti, J.R., Jr. Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J. Biol. Chem. 1997, 272, 8071–8076. [Google Scholar] [CrossRef] [Green Version]
- Trombetta, A.; Maggiora, M.; Martinasso, G.; Cotogni, P.; Canuto, R.A.; Muzio, G. Arachidonic and docosahexaenoic acids reduce the growth of A549 human lung-tumor cells increasing lipid peroxidation and PPARs. Chem. Biol. Interact. 2007, 165, 239–250. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Heim, M.; Johnson, J.; Boess, F.; Bendik, I.; Weber, P.; Hunziker, W.; Fluhmann, B. Phytanic acid, a natural peroxisome proliferator-activated receptor (PPAR) agonist, regulates glucose metabolism in rat primary hepatocytes. FASEB J. 2002, 16, 718–720. [Google Scholar] [CrossRef] [Green Version]
- Marion-Letellier, R.; Dechelotte, P.; Iacucci, M.; Ghosh, S. Dietary modulation of peroxisome proliferator-activated receptor gamma. Gut 2009, 58, 586–593. [Google Scholar] [CrossRef]
- Schwab, M.; Reynders, V.; Loitsch, S.; Steinhilber, D.; Stein, J.; Schroder, O. Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NF kappa B signalling. Mol. Immunol. 2007, 44, 3625–3632. [Google Scholar] [CrossRef]
- Wachtershauser, A.; Loitsch, S.M.; Stein, J. PPAR-gamma is selectively upregulated in Caco-2 cells by butyrate. Biochem. Biophys. Res. Commun. 2000, 272, 380–385. [Google Scholar]
- Nepelska, M.; de Wouters, T.; Jacouton, E.; Beguet-Crespel, F.; Lapaque, N.; Dore, J.; Arulampalam, V.; Blottiere, H.M. Commensal gut bacteria modulate phosphorylation-dependent PPARgamma transcriptional activity in human intestinal epithelial cells. Sci. Rep. 2017, 7, 43199. [Google Scholar] [CrossRef] [Green Version]
- Voltan, S.; Martines, D.; Elli, M.; Brun, P.; Longo, S.; Porzionato, A.; Macchi, V.; D’Inca, R.; Scarpa, M.; Palu, G.; et al. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-gamma in the intestinal mucosa. Gastroenterology 2008, 135, 1216–1227. [Google Scholar] [CrossRef]
- Are, A.; Aronsson, L.; Wang, S.; Greicius, G.; Lee, Y.K.; Gustafsson, J.A.; Pettersson, S.; Arulampalam, V. Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1943–1948. [Google Scholar] [CrossRef] [Green Version]
- Couvigny, B.; de Wouters, T.; Kaci, G.; Jacouton, E.; Delorme, C.; Dore, J.; Renault, P.; Blottiere, H.M.; Guedon, E.; Lapaque, N. Commensal Streptococcus salivarius Modulates PPARgamma Transcriptional Activity in Human Intestinal Epithelial Cells. PLoS ONE 2015, 10, e0125371. [Google Scholar] [CrossRef] [Green Version]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Burgermeister, E.; Schnoebelen, A.; Flament, A.; Benz, J.; Stihle, M.; Gsell, B.; Rufer, A.; Ruf, A.; Kuhn, B.; Marki, H.P.; et al. A novel partial agonist of peroxisome proliferator-activated receptor-gamma (PPARgamma) recruits PPARgamma-coactivator-1alpha, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol. Endocrinol. 2006, 20, 809–830. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Cao, X.; Hu, X.; Li, X.; Shi, X.; Wang, H.; Peng, C.; Li, J.; Li, J.; Li, Q.; et al. CMHX008, a PPARgamma partial agonist, enhances insulin sensitivity with minor influences on bone loss. Genes Dis. 2018, 5, 290–299. [Google Scholar] [CrossRef]
- Henke, B.R. Peroxisome proliferator-activated receptor alpha/gamma dual agonists for the treatment of type 2 diabetes. J. Med. Chem. 2004, 47, 4118–4127. [Google Scholar] [CrossRef]
- Kim, S.H.; Hong, S.H.; Park, Y.J.; Sung, J.H.; Suh, W.; Lee, K.W.; Jung, K.; Lim, C.; Kim, J.H.; Kim, H.; et al. MD001, a Novel Peroxisome Proliferator-activated Receptor alpha/gamma Agonist, Improves Glucose and Lipid Metabolism. Sci. Rep. 2019, 9, 1656. [Google Scholar] [CrossRef]
- Jeong, H.W.; Lee, J.W.; Kim, W.S.; Choe, S.S.; Kim, K.H.; Park, H.S.; Shin, H.J.; Lee, G.Y.; Shin, D.; Lee, H.; et al. A newly identified CG301269 improves lipid and glucose metabolism without body weight gain through activation of peroxisome proliferator-activated receptor alpha and gamma. Diabetes 2011, 60, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, L.L.; Liu, H.Y.; Zhou, X.B.; Cao, Y.L.; Li, S. C333H, a novel PPARalpha/gamma dual agonist, has beneficial effects on insulin resistance and lipid metabolism. Acta Pharmacol. Sin. 2006, 27, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Park, J.Y.; Lee, H.J.; Kim, D.H.; Park, D.; Jeong, H.O.; Park, C.H.; Chun, P.; Moon, H.R.; Chung, H.Y. Potent anti-diabetic effects of MHY908, a newly synthesized PPAR alpha/gamma dual agonist in db/db mice. PLoS ONE 2013, 8, e78815. [Google Scholar] [CrossRef] [Green Version]
- Waites, C.R.; Dominick, M.A.; Sanderson, T.P.; Schilling, B.E. Nonclinical safety evaluation of muraglitazar, a novel PPARalpha/gamma agonist. Toxicol. Sci. 2007, 100, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, R.; Vikramadithyan, R.K.; Misra, P.; Hiriyan, J.; Raichur, S.; Damarla, R.K.; Gershome, C.; Suresh, J.; Rajagopalan, R. Ragaglitazar: A novel PPAR alpha PPAR gamma agonist with potent lipid-lowering and insulin-sensitizing efficacy in animal models. Br. J. Pharmacol. 2003, 140, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Masternak, M.M.; Bartke, A. PPARs in Calorie Restricted and Genetically Long-Lived Mice. PPAR Res. 2007, 2007, 28436. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.B.; Wu, X.L.; Ke, B.; Huang, Y.J.; Chen, S.Q.; Su, Y.Q.; Qin, J. Effects of caloric restriction on peroxisome proliferator-activated receptors and positive transcription elongation factor b expression in obese rats. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4369–4378. [Google Scholar]
- Masternak, M.M.; Al-Regaiey, K.A.; Del Rosario Lim, M.M.; Jimenez-Ortega, V.; Panici, J.A.; Bonkowski, M.S.; Kopchick, J.J.; Wang, Z.; Bartke, A. Caloric restriction and growth hormone receptor knockout: Effects on expression of genes involved in insulin action in the heart. Exp. Gerontol. 2006, 41, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Masternak, M.M.; Al-Regaiey, K.A.; Del Rosario Lim, M.M.; Jimenez-Ortega, V.; Panici, J.A.; Bonkowski, M.S.; Kopchick, J.J.; Bartke, A. Effects of caloric restriction and growth hormone resistance on the expression level of peroxisome proliferator-activated receptors superfamily in liver of normal and long-lived growth hormone receptor/binding protein knockout mice. J. Gerontol. Biol. Sci. Med. Sci. 2005, 60, 1394–1398. [Google Scholar] [CrossRef]
- Masternak, M.M.; Al-Regaiey, K.; Bonkowski, M.S.; Panici, J.; Sun, L.; Wang, J.; Przybylski, G.K.; Bartke, A. Divergent effects of caloric restriction on gene expression in normal and long-lived mice. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 784–788. [Google Scholar] [CrossRef] [Green Version]
- Masternak, M.M.; Al-Regaiey, K.A.; Del Rosario Lim, M.M.; Bonkowski, M.S.; Panici, J.A.; Przybylski, G.K.; Bartke, A. Caloric restriction results in decreased expression of peroxisome proliferator-activated receptor superfamily in muscle of normal and long-lived growth hormone receptor/binding protein knockout mice. J. Gerontol. Biol. Sci. Med. Sci. 2005, 60, 1238–1245. [Google Scholar] [CrossRef]
- Poynter, M.E.; Daynes, R.A. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998, 273, 32833–32841. [Google Scholar]
- Duszka, K.; Ellero-Simatos, S.; Ow, G.S.; Defernez, M.; Paramalingam, E.; Tett, A.; Ying, S.; Konig, J.; Narbad, A.; Kuznetsov, V.A.; et al. Complementary intestinal mucosa and microbiota responses to caloric restriction. Sci. Rep. 2018, 8, 11338. [Google Scholar] [CrossRef]
- Dhahbi, J.M.; Tsuchiya, T.; Kim, H.J.; Mote, P.L.; Spindler, S.R. Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. J. Gerontol. Biol. Sci. Med. Sci. 2006, 61, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.; Park, S.; Yu, B.P.; Chung, H.Y. Modulation of PPAR in aging, inflammation, and calorie restriction. J. Gerontol. Biol. Sci. Med. Sci. 2004, 59, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.C.; Apte, U.; Anderson, S.P.; Limaye, P.; Yoon, L.; Latendresse, J.; Dunn, C.; Everitt, J.I.; Voss, K.A.; Swanson, C.; et al. Mimetics of caloric restriction include agonists of lipid-activated nuclear receptors. J. Biol. Chem. 2004, 279, 46204–46212. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Liang, Y.; He, Q.; Yao, R.; Bao, W.; Bao, L.; Wang, Y.; Wang, Z. Current models of mammalian target of rapamycin complex 1 (mTORC1) activation by growth factors and amino acids. Int. J. Mol. Sci. 2014, 15, 20753–20769. [Google Scholar] [CrossRef] [Green Version]
- Tatebe, H.; Shiozaki, K. Evolutionary Conservation of the Components in the TOR Signaling Pathways. Biomolecules 2017, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Yamagata, K.; Sanders, L.K.; Kaufmann, W.E.; Yee, W.; Barnes, C.A.; Nathans, D.; Worley, P.F. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 1994, 269, 16333–16339. [Google Scholar]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar]
- Wang, X.; Proud, C.G. The mTOR pathway in the control of protein synthesis. Physiology 2006, 21, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef]
- Kwiatkowski, D.J. Tuberous sclerosis: From tubers to mTOR. Ann. Hum. Genet. 2003, 67, 87–96. [Google Scholar]
- Miyazaki, M.; McCarthy, J.J.; Esser, K.A. Insulin like growth factor-1-induced phosphorylation and altered distribution of tuberous sclerosis complex (TSC)1/TSC2 in C2C12 myotubes. FEBS J. 2010, 277, 2180–2191. [Google Scholar] [CrossRef] [Green Version]
- Dan, H.C.; Sun, M.; Yang, L.; Feldman, R.I.; Sui, X.M.; Ou, C.C.; Nellist, M.; Yeung, R.S.; Halley, D.J.; Nicosia, S.V.; et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 2002, 277, 35364–35370. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, Y.H.; Wu, X.N.; Wu, S.Q.; Lu, B.J.; Dong, M.Q.; Zhang, H.; Sun, P.; Lin, S.C.; Guan, K.L.; et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Arsham, A.M.; Neufeld, T.P. Thinking globally and acting locally with TOR. Curr. Opin. Cell Biol. 2006, 18, 589–597. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Reiling, J.H.; Hafen, E. The hypoxia-induced paralogs Scylla and CharybDis. inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004, 18, 2879–2892. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Sabatini, D.M. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 2005, 280, 39505–39509. [Google Scholar] [CrossRef] [Green Version]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Dann, S.G.; Thomas, G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett. 2006, 580, 2821–2829. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [Green Version]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar]
- Kim, J.E.; Chen, J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar]
- Zhang, H.H.; Huang, J.; Duvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.T.; Ducker, G.S.; Barczak, A.J.; Barbeau, R.; Erle, D.J.; Shokat, K.M. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc. Natl. Acad. Sci. USA 2011, 108, 15201–15206. [Google Scholar] [CrossRef] [Green Version]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.H.; et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Corradetti, M.N.; Guan, K.L. Upstream of the mammalian target of rapamycin: Do all roads pass through mTOR? Oncogene 2006, 25, 6347–6360. [Google Scholar] [CrossRef] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.; Blenis, J.; Pende, M.; Sonenberg, N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Richardson, C.J.; Broenstrup, M.; Fingar, D.C.; Julich, K.; Ballif, B.A.; Gygi, S.; Blenis, J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr. Biol. 2004, 14, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Blenis, J.; Kuo, C.J.; Erikson, R.L. Identification of a ribosomal protein S6 kinase regulated by transformation and growth-promoting stimuli. J. Biol. Chem. 1987, 262, 14373–14376. [Google Scholar]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, G.; Gerst, F.; Michael, D.; Berchtold, S.; Friedrich, B.; Strutz-Seebohm, N.; Lang, F.; Haring, H.U.; Ullrich, S. Regulation of forkhead box O1 (FOXO1) by protein kinase B and glucocorticoids: Different mechanisms of induction of beta cell death in vitro. Diabetologia 2013, 56, 1587–1595. [Google Scholar] [CrossRef]
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and acetylation modifications of FOXO3a: Independently or synergistically? Oncol. Lett. 2017, 13, 2867–2872. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Powers, R.W., 3rd; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [Green Version]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Muller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Medvedik, O.; Lamming, D.W.; Kim, K.D.; Sinclair, D.A. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007, 5, e261. [Google Scholar] [CrossRef] [Green Version]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [Green Version]
- Toth, M.L.; Sigmond, T.; Borsos, E.; Barna, J.; Erdelyi, P.; Takacs-Vellai, K.; Orosz, L.; Kovacs, A.L.; Csikos, G.; Sass, M.; et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Taubert, S.; Crawford, D.; Libina, N.; Lee, S.J.; Kenyon, C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 2007, 6, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008, 8, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Cota, D.; Matter, E.K.; Woods, S.C.; Seeley, R.J. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J. Neurosci. 2008, 28, 7202–7208. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef]
- Kim, K.; Pyo, S.; Um, S.H. S6 kinase 2 deficiency enhances ketone body production and increases peroxisome proliferator-activated receptor alpha activity in the liver. Hepatology 2012, 55, 1727–1737. [Google Scholar] [CrossRef]
- Gebert, N.; Cheng, C.W.; Kirkpatrick, J.M.; Di Fraia, D.; Yun, J.; Schadel, P.; Pace, S.; Garside, G.B.; Werz, O.; Rudolph, K.L.; et al. Region-Specific Proteome Changes of the Intestinal Epithelium during Aging and Dietary Restriction. Cell Rep. 2020, 31, 107565. [Google Scholar] [CrossRef]
- Pentinmikko, N.; Iqbal, S.; Mana, M.; Andersson, S.; Cognetta, A.B., 3rd; Suciu, R.M.; Roper, J.; Luopajarvi, K.; Markelin, E.; Gopalakrishnan, S.; et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 2019, 571, 398–402. [Google Scholar] [CrossRef]
- Okuda, Y.; Kawai, K.; Yamashita, K. Age-related change in ketone body metabolism: Diminished glucagon effect on ketogenesis in adult rats. Endocrinology 1987, 120, 2152–2157. [Google Scholar] [CrossRef]
- Sanguino, E.; Ramon, M.; Michalik, L.; Wahli, W.; Alegret, M.; Sanchez, R.M.; Vazquez-Carrera, M.; Laguna, J.C. Lack of hypotriglyceridemic effect of gemfibrozil as a consequence of age-related changes in rat liver PPARalpha. Biochem. Pharmacol. 2004, 67, 157–166. [Google Scholar] [CrossRef]
- Sastre, J.; Pallardo, F.V.; Pla, R.; Pellin, A.; Juan, G.; O’Connor, J.E.; Estrela, J.M.; Miquel, J.; Vina, J. Aging of the liver: Age-associated mitochondrial damage in intact hepatocytes. Hepatology 1996, 24, 1199–1205. [Google Scholar] [CrossRef]
- Jiao, M.; Ren, F.; Zhou, L.; Zhang, X.; Zhang, L.; Wen, T.; Wei, L.; Wang, X.; Shi, H.; Bai, L.; et al. Peroxisome proliferator-activated receptor alpha activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 2014, 5, e1397. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Kim, K.H.; Moore, D.D. Regulation of Liver Energy Balance by the Nuclear Receptors Farnesoid X Receptor and Peroxisome Proliferator Activated Receptor alpha. Dig. Dis. 2017, 35, 203–209. [Google Scholar] [CrossRef]
- Sun, X.; Ritzenthaler, J.D.; Zhong, X.; Zheng, Y.; Roman, J.; Han, S. Nicotine stimulates PPARbeta/delta expression in human lung carcinoma cells through activation of PI3K/mTOR and suppression of AP-2alpha. Cancer Res. 2009, 69, 6445–6453. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Ritzenthaler, J.D.; Zheng, Y.; Roman, J. PPARbeta/delta agonist stimulates human lung carcinoma cell growth through inhibition of PTEN expression: The involvement of PI3K and NF-kappaB signals. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1238–L1249. [Google Scholar] [CrossRef]
- Foster, D.A. Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol. Metab. 2013, 24, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Park, J.; Lee, H.W.; Lee, Y.S.; Kim, J.B. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem. Biophys. Res. Commun. 2004, 321, 942–948. [Google Scholar] [CrossRef]
- Gagnon, A.; Lau, S.; Sorisky, A. Rapamycin-sensitive phase of 3T3-L1 preadipocyte differentiation after clonal expansion. J. Cell Physiol. 2001, 189, 14–22. [Google Scholar] [CrossRef]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Ruegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef]
- Bell, A.; Grunder, L.; Sorisky, A. Rapamycin inhibits human adipocyte differentiation in primary culture. Obes. Res. 2000, 8, 249–254. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Masuda, K.; Frigerio, F.; Le Bacquer, O.; Um, S.H.; Gandin, V.; Topisirovic, I.; Sonenberg, N.; Thomas, G.; Kozma, S.C. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell 2010, 18, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, P.G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brule, S.; Turcotte, V.; Cote, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARgamma-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Wright, H.M.; Wright, M.; Spiegelman, B.M. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4333–4337. [Google Scholar] [CrossRef] [Green Version]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef]
- Huffman, T.A.; Mothe-Satney, I.; Lawrence, J.C., Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. USA 2002, 99, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.K.; Lee, M.Y.; Kim, J.W.; Kim, M.; Moon, J.S.; Lee, Y.J.; Ahn, Y.H.; Kim, K.S. Lipin1 is a key factor for the maturation and maintenance of adipocytes in the regulatory network with CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma 2. J. Biol. Chem. 2008, 283, 34896–34906. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.E.; Bae, E.; Jeong, D.Y.; Kim, M.J.; Jin, W.J.; Park, S.W.; Han, G.S.; Carman, G.M.; Koh, E.; Kim, K.S. Lipin1 regulates PPARgamma transcriptional activity. Biochem. J. 2013, 453, 49–60. [Google Scholar] [CrossRef]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C., Jr.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [Green Version]
- San, Y.Z.; Liu, Y.; Zhang, Y.; Shi, P.P.; Zhu, Y.L. Peroxisome proliferator-activated receptor-gamma agonist inhibits the mammalian target of rapamycin signaling pathway and has a protective effect in a rat model of status epilepticus. Mol. Med. Rep. 2015, 12, 1877–1883. [Google Scholar] [CrossRef] [Green Version]
- Purnell, P.; Teepper, C.; Liu, S.; Cardiff, R.; Gregg, J. Induction of PPAR gamma signaling and autophagy as a mechanism mediating acquired rapamycin resistance in breast cancer models. Cancer Res. 2008, 68. [Google Scholar]
- Assumpcao, J.A.F.; Magalhaes, K.G.; Correa, J.R. The role of ppargamma and autophagy in ros production, lipid droplets biogenesis and its involvement with colorectal cancer cells modulation. Cancer Cell Int. 2017, 17, 82. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Chang, Y.C.; Maurizi, M.R. 4-O-carboxymethyl ascochlorin causes ER stress and induced autophagy in human hepatocellular carcinoma cells. J. Biol. Chem. 2012, 287, 15661–15671. [Google Scholar] [CrossRef] [Green Version]
- Pellerito, O.; Notaro, A.; Sabella, S.; De Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARgamma down-regulation. Apoptosis 2014, 19, 1029–1042. [Google Scholar] [CrossRef]
- Weng, J.R.; Bai, L.Y.; Chiu, C.F.; Hu, J.L.; Chiu, S.J.; Wu, C.Y. Cucurbitane Triterpenoid from Momordica charantia Induces Apoptosis and Autophagy in Breast Cancer Cells, in Part, through Peroxisome Proliferator-Activated Receptor gamma Activation. Evid. Based Complement. Altern. Med. 2013, 2013, 935675. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, W.; Liang, B.; Casimiro, M.C.; Whitaker-Menezes, D.; Wang, M.; Lisanti, M.P.; Lanza-Jacoby, S.; Pestell, R.G.; Wang, C. PPARgamma activation induces autophagy in breast cancer cells. Int. J. Biochem. Cell Biol. 2009, 41, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Yang, X.; Chen, T.; Xi, Z.; Jiang, X. The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014, 21, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Cerquetti, L.; Sampaoli, C.; Amendola, D.; Bucci, B.; Masuelli, L.; Marchese, R.; Misiti, S.; De Venanzi, A.; Poggi, M.; Toscano, V.; et al. Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp. Cell Res. 2011, 317, 1397–1410. [Google Scholar] [CrossRef]
- Teresi, R.E.; Waite, K.A. PPARgamma, PTEN, and the Fight against Cancer. PPAR Res. 2008, 2008, 932632. [Google Scholar] [CrossRef] [Green Version]
- Patel, L.; Pass, I.; Coxon, P.; Downes, C.P.; Smith, S.A.; Macphee, C.H. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr. Biol. 2001, 11, 764–768. [Google Scholar]
- Zhang, W.; Wu, N.; Li, Z.; Wang, L.; Jin, J.; Zha, X.L. PPARgamma activator rosiglitazone inhibits cell migration via upregulation of PTEN in human hepatocarcinoma cell line BEL-7404. Cancer Biol. Ther. 2006, 5, 1008–1014. [Google Scholar]
- Lin, C.F.; Young, K.C.; Bai, C.H.; Yu, B.C.; Ma, C.T.; Chien, Y.C.; Chiang, C.L.; Liao, C.S.; Lai, H.W.; Tsao, C.W. Rosiglitazone regulates anti-inflammation and growth inhibition via PTEN. Biomed. Res. Int. 2014, 2014, 787924. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yin, Y.; Hou, G.; Kang, J.; Wang, Q. Peroxisome Proliferator-Activated Receptor (PPARgamma) Plays a Protective Role in Cigarette Smoking-Induced Inflammation via AMP-Activated Protein Kinase (AMPK) Signaling. Med. Sci. Monit. 2018, 24, 5168–5177. [Google Scholar] [CrossRef]
- He, G.; Sung, Y.M.; Digiovanni, J.; Fischer, S.M. Thiazolidinediones inhibit insulin-like growth factor-i-induced activation of p70S6 kinase and suppress insulin-like growth factor-I tumor-promoting activity. Cancer Res. 2006, 66, 1873–1878. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Lee, H.Y.; Kim, T.G.; Lee, N.H.; Yu, M.K.; Yi, H.K. PPARgamma Maintains Homeostasis through Autophagy Regulation in Dental Pulp. J. Dent. Res. 2015, 94, 729–737. [Google Scholar] [CrossRef]
- Xu, F.; Li, J.; Ni, W.; Shen, Y.W.; Zhang, X.P. Peroxisome proliferator-activated receptor-gamma agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS ONE 2013, 8, e55080. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, Q.; Yang, X.; Wang, L. PPAR-gamma agonist rosiglitazone reduces autophagy and promotes functional recovery in experimental traumaticspinal cord injury. Neurosci. Lett. 2017, 650, 89–96. [Google Scholar] [CrossRef]
- Mahmood, D.F.; Jguirim-Souissi, I.; Khadija, E.H.; Blondeau, N.; Diderot, V.; Amrani, S.; Slimane, M.N.; Syrovets, T.; Simmet, T.; Rouis, M. Peroxisome proliferator-activated receptor gamma induces apoptosis and inhibits autophagy of human monocyte-derived macrophages via induction of cathepsin L: Potential role in atherosclerosis. J. Biol. Chem. 2011, 286, 28858–28866. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef]
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, S.; Hawley, S.A.; Green, K.A.; Saner, N.; Mustard, K.J.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: Synergistic effects of Ca2+ and AMP. Biochem. J. 2010, 426, 109–118. [Google Scholar] [CrossRef]
- Winder, W.W.; Hardie, D.G. AMP-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol. 1999, 277, E1–E10. [Google Scholar] [CrossRef]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef]
- Chen, S.; Murphy, J.; Toth, R.; Campbell, D.G.; Morrice, N.A.; Mackintosh, C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem. J. 2008, 409, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, K.M.; Chen, S.; Harthill, J.E.; Ibrahim, A.F.; Toth, R.; Morrice, N.A.; Vandermoere, F.; Moorhead, G.B.; Hardie, D.G.; MacKintosh, C. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem. J. 2007, 407, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef]
- Davies, S.P.; Sim, A.T.; Hardie, D.G. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 1990, 187, 183–190. [Google Scholar]
- Davies, S.P.; Carling, D.; Munday, M.R.; Hardie, D.G. Diurnal rhythm of phosphorylation of rat liver acetyl-CoA carboxylase by the AMP-activated protein kinase, demonstrated using freeze-clamping. Effects of high fat diets. Eur. J. Biochem. 1992, 203, 615–623. [Google Scholar]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Seefeld, K.; Witters, L.A.; Coleman, R.A. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: Evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 1999, 338 Pt 3, 783–791. [Google Scholar]
- Clarke, P.R.; Hardie, D.G. Regulation of HMG-CoA reductase: Identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990, 9, 2439–2446. [Google Scholar]
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. USA 2009, 106, 17781–17786. [Google Scholar] [CrossRef] [Green Version]
- Dufer, M.; Noack, K.; Krippeit-Drews, P.; Drews, G. Activation of the AMP-activated protein kinase enhances glucose-stimulated insulin secretion in mouse beta-cells. Islets 2010, 2, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Roach, W.G.; Keller, S.R.; Lane, W.S.; Lienhard, G.E. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 9187–9195. [Google Scholar] [CrossRef] [Green Version]
- Marsin, A.S.; Bouzin, C.; Bertrand, L.; Hue, L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 2002, 277, 30778–30783. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, S.B.; Nielsen, J.N.; Birk, J.B.; Olsen, G.S.; Viollet, B.; Andreelli, F.; Schjerling, P.; Vaulont, S.; Hardie, D.G.; Hansen, B.F.; et al. The alpha2-5′ AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 2004, 53, 3074–3081. [Google Scholar]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, I.; Lenzner, C.; Gourdon, L.; Vaulont, S.; Kahn, A.; Viollet, B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes 2001, 50, 1515–1521. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Seo, W.Y.; Song, K.H.; Chanda, D.; Kim, Y.D.; Kim, D.K.; Lee, M.W.; Ryu, D.; Kim, Y.H.; Noh, J.R.; et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010, 285, 32182–32191. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferre, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Andersson, U.; Filipsson, K.; Abbott, C.R.; Woods, A.; Smith, K.; Bloom, S.R.; Carling, D.; Small, C.J. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004, 279, 12005–12008. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Liu, P.; Xie, X. AMPK and Cancer. Exp. Suppl. 2016, 107, 203–226. [Google Scholar] [CrossRef]
- Umezawa, S.; Higurashi, T.; Nakajima, A. AMPK: Therapeutic Target for Diabetes and Cancer Prevention. Curr. Pharm. Des. 2017, 23, 3629–3644. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Funakoshi, M.; Tsuda, M.; Muramatsu, K.; Hatsuda, H.; Morishita, S.; Aigaki, T. A gain-of-function screen identifies wdb and lkb1 as lifespan-extending genes in Drosophila. Biochem. Biophys. Res. Commun. 2011, 405, 667–672. [Google Scholar] [CrossRef]
- Stenesen, D.; Suh, J.M.; Seo, J.; Yu, K.; Lee, K.S.; Kim, J.S.; Min, K.J.; Graff, J.M. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 2013, 17, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Carling, D.; Carlson, M. The AMP-activated/SNF1 protein kinase subfamily: Metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 1998, 67, 821–855. [Google Scholar] [CrossRef]
- Viollet, B.; Andreelli, F. AMP-activated protein kinase and metabolic control. Handb. Exp. Pharmacol. 2011, 303–330. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.A.; Vanden Heuvel, J.P. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Miller, E.J.; Ninomiya-Tsuji, J.; Russell, R.R., 3rd; Young, L.H. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ. Res. 2005, 97, 872–879. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, K.; Konrad, D.; Klip, A.; Marette, A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. FASEB J. 2003, 17, 1658–1665. [Google Scholar] [CrossRef]
- Hunter, R.W.; Treebak, J.T.; Wojtaszewski, J.F.; Sakamoto, K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 2011, 60, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Kim, M.; Park, H.S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.S.; Oh, G.T.; et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [Green Version]
- Gan, Z.; Burkart-Hartman, E.M.; Han, D.H.; Finck, B.; Leone, T.C.; Smith, E.Y.; Ayala, J.E.; Holloszy, J.; Kelly, D.P. The nuclear receptor PPARbeta/delta programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes Dev. 2011, 25, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Bai, F.; Liu, Y.; Tu, T.; Li, B.; Xiao, Y.; Ma, Y.; Qin, F.; Xie, J.; Zhou, S.; Liu, Q. Metformin regulates lipid metabolism in a canine model of atrial fibrillation through AMPK/PPAR-alpha/VLCAD pathway. Lipids Health Dis. 2019, 18, 109. [Google Scholar] [CrossRef] [Green Version]
- Joly, E.; Roduit, R.; Peyot, M.L.; Habinowski, S.A.; Ruderman, N.B.; Witters, L.A.; Prentki, M. Glucose represses PPARalpha gene expression via AMP-activated protein kinase but not via p38 mitogen-activated protein kinase in the pancreatic beta-cell. J. Diabetes 2009, 1, 263–272. [Google Scholar] [CrossRef]
- Ravnskjaer, K.; Boergesen, M.; Dalgaard, L.T.; Mandrup, S. Glucose-induced repression of PPARalpha gene expression in pancreatic beta-cells involves PP2A activation and AMPK inactivation. J. Mol. Endocrinol. 2006, 36, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Bronner, M.; Hertz, R.; Bar-Tana, J. Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem. J. 2004, 384, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Sozio, M.S.; Lu, C.; Zeng, Y.; Liangpunsakul, S.; Crabb, D.W. Activated AMPK inhibits PPAR-{alpha} and PPAR-{gamma} transcriptional activity in hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G739–G747. [Google Scholar] [CrossRef] [Green Version]
- Chanda, D.; Lee, C.H.; Kim, Y.H.; Noh, J.R.; Kim, D.K.; Park, J.H.; Hwang, J.H.; Lee, M.R.; Jeong, K.H.; Lee, I.K.; et al. Fenofibrate differentially regulates plasminogen activator inhibitor-1 gene expression via adenosine monophosphate-activated protein kinase-dependent induction of orphan nuclear receptor small heterodimer partner. Hepatology 2009, 50, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Liangpunsakul, S.; Wou, S.E.; Wineinger, K.D.; Zeng, Y.; Cyganek, I.; Jayaram, H.N.; Crabb, D.W. Effects of WY-14,643 on the phosphorylation and activation of AMP-dependent protein kinase. Arch. Biochem. Biophys. 2009, 485, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Rimando, A.M.; Mathews, S.T. AMPK activation by pterostilbene contributes to suppression of hepatic gluconeogenic gene expression and glucose production in H4IIE cells. Biochem. Biophys. Res. Commun. 2018, 498, 640–645. [Google Scholar] [CrossRef]
- Murakami, H.; Murakami, R.; Kambe, F.; Cao, X.; Takahashi, R.; Asai, T.; Hirai, T.; Numaguchi, Y.; Okumura, K.; Seo, H.; et al. Fenofibrate activates AMPK and increases eNOS phosphorylation in HUVEC. Biochem. Biophys. Res. Commun. 2006, 341, 973–978. [Google Scholar] [CrossRef]
- Okayasu, T.; Tomizawa, A.; Suzuki, K.; Manaka, K.; Hattori, Y. PPARalpha activators upregulate eNOS activity and inhibit cytokine-induced NF-kappaB activation through AMP-activated protein kinase activation. Life Sci. 2008, 82, 884–891. [Google Scholar] [CrossRef]
- Manio, M.C.; Inoue, K.; Fujitani, M.; Matsumura, S.; Fushiki, T. Combined pharmacological activation of AMPK and PPARdelta potentiates the effects of exercise in trained mice. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Lamontagne, J.; Pepin, E.; Peyot, M.L.; Joly, E.; Ruderman, N.B.; Poitout, V.; Madiraju, S.R.; Nolan, C.J.; Prentki, M. Pioglitazone acutely reduces insulin secretion and causes metabolic deceleration of the pancreatic beta-cell at submaximal glucose concentrations. Endocrinology 2009, 150, 3465–3474. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.G.; Logan, P.J.; Ewart, M.A.; Reihill, J.A.; Ritchie, S.A.; Connell, J.M.; Cleland, S.J.; Salt, I.P. Rosiglitazone stimulates nitric oxide synthesis in human aortic endothelial cells via AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 11210–11217. [Google Scholar] [CrossRef] [Green Version]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Zhu, X.; Lin, W.; Zhou, Y.; Cai, W.; Qiu, L. Interactions of TLR4 and PPARgamma, Dependent on AMPK Signalling Pathway Contribute to Anti-Inflammatory Effects of Vaccariae Hypaphorine in Endothelial Cells. Cell Physiol. Biochem. 2017, 42, 1227–1239. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, L.; Shao, L.; Qian, L.; Fu, X.; Li, G.; Luo, T.; Gu, Y.; Li, F.; Li, J.; et al. Troglitazone acutely activates AMP-activated protein kinase and inhibits insulin secretion from beta cells. Life Sci. 2007, 81, 160–165. [Google Scholar] [CrossRef]
- LeBrasseur, N.K.; Kelly, M.; Tsao, T.S.; Farmer, S.R.; Saha, A.K.; Ruderman, N.B.; Tomas, E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E175–E181. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Liang, X.; Rogers, C.Q.; Rideout, D.; You, M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G364–G374. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.K.; Avilucea, P.R.; Ye, J.M.; Assifi, M.M.; Kraegen, E.W.; Ruderman, N.B. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem. Biophys. Res. Commun. 2004, 314, 580–585. [Google Scholar] [CrossRef]
- Ye, J.M.; Dzamko, N.; Hoy, A.J.; Iglesias, M.A.; Kemp, B.; Kraegen, E. Rosiglitazone treatment enhances acute AMP-activated protein kinase-mediated muscle and adipose tissue glucose uptake in high-fat-fed rats. Diabetes 2006, 55, 2797–2804. [Google Scholar] [CrossRef] [Green Version]
- Coletta, D.K.; Sriwijitkamol, A.; Wajcberg, E.; Tantiwong, P.; Li, M.; Prentki, M.; Madiraju, M.; Jenkinson, C.P.; Cersosimo, E.; Musi, N.; et al. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: A randomised trial. Diabetologia 2009, 52, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Su, G.; Brown, S.N.; Chen, L.; Ren, J.; Zhao, P. Peroxisome proliferator-activated receptors gamma and alpha agonists stimulate cardiac glucose uptake via activation of AMP-activated protein kinase. J. Nutr. Biochem. 2010, 21, 621–626. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; Group, L.S. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Zhang, F.; Dey, D.; Branstrom, R.; Forsberg, L.; Lu, M.; Zhang, Q.; Sjoholm, A. BLX-1002, a novel thiazolidinedione with no PPAR affinity, stimulates AMP-activated protein kinase activity, raises cytosolic Ca2+, and enhances glucose-stimulated insulin secretion in a PI3K-dependent manner. Am. J. Physiol. Cell Physiol. 2009, 296, C346–C354. [Google Scholar] [CrossRef]
- Chang, T.J.; Chen, W.P.; Yang, C.; Lu, P.H.; Liang, Y.C.; Su, M.J.; Lee, S.C.; Chuang, L.M. Serine-385 phosphorylation of inwardly rectifying K+ channel subunit (Kir6.2) by AMP-dependent protein kinase plays a key role in rosiglitazone-induced closure of the K(ATP) channel and insulin secretion in rats. Diabetologia 2009, 52, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Piscitello, D.; Varshney, D.; Lilla, S.; Vizioli, M.G.; Reid, C.; Gorbunova, V.; Seluanov, A.; Gillespie, D.A.; Adams, P.D. AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11. Oncogene 2018, 37, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Song, W.; Zhang, F.; Yan, J.; Yang, Q. Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway. Oncogene 2013, 32, 1943–1949. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol. Cell 2015, 57, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Rowland, A.F.; Fazakerley, D.J.; James, D.E. Mapping insulin/GLUT4 circuitry. Traffic 2011, 12, 672–681. [Google Scholar] [CrossRef]
- Bevan, P. Insulin signalling. J. Cell Sci. 2001, 114, 1429–1430. [Google Scholar]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.H.; Sul, H.S. Insulin signaling in fatty acid and fat synthesis: A transcriptional perspective. Curr. Opin. Pharmacol. 2010, 10, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Weindruch, R. The retardation of aging by caloric restriction: Studies in rodents and primates. Toxicol. Pathol. 1996, 24, 742–745. [Google Scholar] [CrossRef]
- Shimokawa, I.; Komatsu, T.; Hayashi, N.; Kim, S.E.; Kawata, T.; Park, S.; Hayashi, H.; Yamaza, H.; Chiba, T.; Mori, R. The life-extending effect of dietary restriction requires Foxo3 in mice. Aging Cell 2015, 14, 707–709. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, M.H.; Lee, E.K.; Choi, Y.J.; Chung, K.W.; Moon, K.M.; Kim, M.J.; An, H.J.; Park, J.W.; Kim, N.D.; et al. The roles of FoxOs in modulation of aging by calorie restriction. Biogerontology 2015, 16, 1–14. [Google Scholar] [CrossRef]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Dosch, J.; Meissner, U.; Rascher, W. Prolonged lifespan by defective insulin signalling? Eur. J. Endocrinol. 2003, 148, 489–490. [Google Scholar] [CrossRef] [Green Version]
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 2007, 317, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Selman, C.; Lingard, S.; Choudhury, A.I.; Batterham, R.L.; Claret, M.; Clements, M.; Ramadani, F.; Okkenhaug, K.; Schuster, E.; Blanc, E.; et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008, 22, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Coschigano, K.T.; Holland, A.N.; Riders, M.E.; List, E.O.; Flyvbjerg, A.; Kopchick, J.J. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology 2003, 144, 3799–3810. [Google Scholar] [CrossRef] [Green Version]
- Bonkowski, M.S.; Rocha, J.S.; Masternak, M.M.; Al Regaiey, K.A.; Bartke, A. Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc. Natl. Acad. Sci. USA 2006, 103, 7901–7905. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Erol, A. Insulin resistance is an evolutionarily conserved physiological mechanism at the cellular level for protection against increased oxidative stress. Bioessays 2007, 29, 811–818. [Google Scholar] [CrossRef]
- Dobbins, R.L.; Chester, M.W.; Stevenson, B.E.; Daniels, M.B.; Stein, D.T.; McGarry, J.D. A fatty acid-dependent step is critically important for both glucose- and non-glucose-stimulated insulin secretion. J. Clin. Investig. 1998, 101, 2370–2376. [Google Scholar] [CrossRef]
- Gremlich, S.; Nolan, C.; Roduit, R.; Burcelin, R.; Peyot, M.L.; Delghingaro-Augusto, V.; Desvergne, B.; Michalik, L.; Prentki, M.; Wahli, W. Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology 2005, 146, 375–382. [Google Scholar] [CrossRef]
- Ravnskjaer, K.; Boergesen, M.; Rubi, B.; Larsen, J.K.; Nielsen, T.; Fridriksson, J.; Maechler, P.; Mandrup, S. Peroxisome proliferator-activated receptor alpha (PPARalpha) potentiates, whereas PPARgamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology 2005, 146, 3266–3276. [Google Scholar] [CrossRef] [Green Version]
- Holness, M.J.; Smith, N.D.; Greenwood, G.K.; Sugden, M.C. Acute (24 h) activation of peroxisome proliferator-activated receptor-alpha (PPARalpha) reverses high-fat feeding-induced insulin hypersecretion in vivo and in perifused pancreatic islets. J. Endocrinol. 2003, 177, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Lalloyer, F.; Vandewalle, B.; Percevault, F.; Torpier, G.; Kerr-Conte, J.; Oosterveer, M.; Paumelle, R.; Fruchart, J.C.; Kuipers, F.; Pattou, F.; et al. Peroxisome proliferator-activated receptor alpha improves pancreatic adaptation to insulin resistance in obese mice and reduces lipotoxicity in human islets. Diabetes 2006, 55, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia 2011, 54, 339–349. [Google Scholar] [CrossRef]
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; Marz, W.; Tafuri, S.; Wahli, W.; et al. PPARalpha governs glycerol metabolism. J. Clin. Investig. 2004, 114, 94–103. [Google Scholar] [CrossRef]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef]
- Sugden, M.C.; Greenwood, G.K.; Smith, N.D.; Holness, M.J. Peroxisome proliferator-activated receptor-alpha activation during pregnancy attenuates glucose-stimulated insulin hypersecretion in vivo by increasing insulin sensitivity, without impairing pregnancy-induced increases in beta-cell glucose sensing and responsiveness. Endocrinology 2003, 144, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, C.; Frezzati, S.; Romussi, M.; Bertazzoni, A.; Testori, G.P.; Antonini, S.; Paracchi, A. Effects of short-term clofibrate administration on glucose tolerance and insulin secretion in patients with chemical diabetes or hypertriglyceridemia. Metabolism 1977, 26, 129–139. [Google Scholar] [CrossRef]
- Murakami, K.; Nambu, S.; Koh, H.; Kobayashi, M.; Shigeta, Y. Clofibrate enhances the affinity of insulin receptors in non-insulin dependent diabetes mellitus. Br. J. Clin. Pharmacol. 1984, 17, 89–91. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Shigeta, Y.; Hirata, Y.; Omori, Y.; Sakamoto, N.; Nambu, S.; Baba, S. Improvement of glucose tolerance in NIDDM by clofibrate. Randomized double-blind study. Diabetes Care 1988, 11, 495–499. [Google Scholar] [CrossRef]
- Koh, E.H.; Kim, M.S.; Park, J.Y.; Kim, H.S.; Youn, J.Y.; Park, H.S.; Youn, J.H.; Lee, K.U. Peroxisome proliferator-activated receptor (PPAR)-alpha activation prevents diabetes in OLETF rats: Comparison with PPAR-gamma activation. Diabetes 2003, 52, 2331–2337. [Google Scholar] [CrossRef] [Green Version]
- Guerre-Millo, M.; Gervois, P.; Raspe, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem. 2000, 275, 16638–16642. [Google Scholar] [CrossRef] [Green Version]
- Aasum, E.; Belke, D.D.; Severson, D.L.; Riemersma, R.A.; Cooper, M.; Andreassen, M.; Larsen, T.S. Cardiac function and metabolism in Type 2 diabetic mice after treatment with BM 17.0744, a novel PPAR-alpha activator. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H949–H957. [Google Scholar] [CrossRef]
- Park, C.W.; Zhang, Y.; Zhang, X.; Wu, J.; Chen, L.; Cha, D.R.; Su, D.; Hwang, M.T.; Fan, X.; Davis, L.; et al. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006, 69, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Bulmer, K.; Gibbons, G.F.; Knight, B.L.; Holness, M.J. Peroxisome-proliferator-activated receptor-alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem. J. 2002, 364, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Guerre-Millo, M.; Rouault, C.; Poulain, P.; Andre, J.; Poitout, V.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Reach, G.; Staels, B. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes 2001, 50, 2809–2814. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.S.; Ryu, D.; Maida, A.; Wang, X.; Evans, R.M.; Schoonjans, K.; Auwerx, J. Phosphorylation of the nuclear receptor corepressor 1 by protein kinase B switches its corepressor targets in the liver in mice. Hepatology 2015, 62, 1606–1618. [Google Scholar] [CrossRef] [Green Version]
- Roduit, R.; Morin, J.; Masse, F.; Segall, L.; Roche, E.; Newgard, C.B.; Assimacopoulos-Jeannet, F.; Prentki, M. Glucose down-regulates the expression of the peroxisome proliferator-activated receptor-alpha gene in the pancreatic beta-cell. J. Biol. Chem. 2000, 275, 35799–35806. [Google Scholar] [CrossRef] [Green Version]
- Blanquart, C.; Mansouri, R.; Paumelle, R.; Fruchart, J.C.; Staels, B.; Glineur, C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol. Endocrinol. 2004, 18, 1906–1918. [Google Scholar] [CrossRef] [Green Version]
- Hostetler, H.A.; Huang, H.; Kier, A.B.; Schroeder, F. Glucose directly links to lipid metabolism through high affinity interaction with peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2008, 283, 2246–2254. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of lipid peroxidation and PPAR-delta in amplifying glucose-stimulated insulin secretion. Diabetes 2011, 60, 2830–2842. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, J.; Barg, S.; Vallois, D.; Lahiri, S.; Roger, C.; Yessoufou, A.; Pradevand, S.; McDonald, A.; Bonal, C.; Reimann, F.; et al. PPARbeta/delta affects pancreatic beta cell mass and insulin secretion in mice. J. Clin. Investig. 2012, 122, 4105–4117. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Marco, L.; Rodriguez-Calvo, R.; El Kochairi, I.; Palomer, X.; Michalik, L.; Wahli, W.; Vazquez-Carrera, M. Activation of peroxisome proliferator-activated receptor-beta/-delta (PPAR-beta/-delta) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes. Diabetes 2011, 60, 1990–1999. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Marco, L.; Barroso, E.; El Kochairi, I.; Palomer, X.; Michalik, L.; Wahli, W.; Vazquez-Carrera, M. The peroxisome proliferator-activated receptor (PPAR) beta/delta agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia 2012, 55, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Salvado, L.; Barroso, E.; Gomez-Foix, A.M.; Palomer, X.; Michalik, L.; Wahli, W.; Vazquez-Carrera, M. PPARbeta/delta prevents endoplasmic reticulum stress-associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2014, 57, 2126–2135. [Google Scholar] [CrossRef]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [Green Version]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, T.A.; Xiang, A.H.; Peters, R.K.; Kjos, S.L.; Marroquin, A.; Goico, J.; Ochoa, C.; Tan, S.; Berkowitz, K.; Hodis, H.N.; et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk hispanic women. Diabetes 2002, 51, 2796–2803. [Google Scholar] [CrossRef] [Green Version]
- Knowler, W.C.; Hamman, R.F.; Edelstein, S.L.; Barrett-Connor, E.; Ehrmann, D.A.; Walker, E.A.; Fowler, S.E.; Nathan, D.M.; Kahn, S.E.; Diabetes Prevention Program Research, G. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes 2005, 54, 1150–1156. [Google Scholar] [CrossRef]
- Moitra, J.; Mason, M.M.; Olive, M.; Krylov, D.; Gavrilova, O.; Marcus-Samuels, B.; Feigenbaum, L.; Lee, E.; Aoyama, T.; Eckhaus, M.; et al. Life without white fat: A transgenic mouse. Genes Dev. 1998, 12, 3168–3181. [Google Scholar]
- Larsen, T.M.; Toubro, S.; Astrup, A. PPARgamma agonists in the treatment of type II diabetes: Is increased fatness commensurate with long-term efficacy? Int. J. Obes. Relat. Metab. Disord. 2003, 27, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.M.; Doyle, P.J.; Iglesias, M.A.; Watson, D.G.; Cooney, G.J.; Kraegen, E.W. Peroxisome proliferator-activated receptor (PPAR)-alpha activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: Comparison with PPAR-gamma activation. Diabetes 2001, 50, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Collino, M.; Aragno, M.; Castiglia, S.; Miglio, G.; Tomasinelli, C.; Boccuzzi, G.; Thiemermann, C.; Fantozzi, R. Pioglitazone improves lipid and insulin levels in overweight rats on a high cholesterol and fructose diet by decreasing hepatic inflammation. Br. J. Pharmacol. 2010, 160, 1892–1902. [Google Scholar] [CrossRef] [Green Version]
- Iwata, M.; Haruta, T.; Usui, I.; Takata, Y.; Takano, A.; Uno, T.; Kawahara, J.; Ueno, E.; Sasaoka, T.; Ishibashi, O.; et al. Pioglitazone ameliorates tumor necrosis factor-alpha-induced insulin resistance by a mechanism independent of adipogenic activity of peroxisome proliferator-activated receptor-gamma. Diabetes 2001, 50, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Smith, U.; Gogg, S.; Johansson, A.; Olausson, T.; Rotter, V.; Svalstedt, B. Thiazolidinediones (PPARgamma agonists) but not PPARalpha agonists increase IRS-2 gene expression in 3T3-L1 and human adipocytes. FASEB J. 2001, 15, 215–220. [Google Scholar] [CrossRef]
- Rieusset, J.; Auwerx, J.; Vidal, H. Regulation of gene expression by activation of the peroxisome proliferator-activated receptor gamma with rosiglitazone (BRL 49653) in human adipocytes. Biochem. Biophys. Res. Commun. 1999, 265, 265–271. [Google Scholar] [CrossRef]
- Baumann, C.A.; Chokshi, N.; Saltiel, A.R.; Ribon, V. Cloning and characterization of a functional peroxisome proliferator activator receptor-gamma-responsive element in the promoter of the CAP gene. J. Biol. Chem. 2000, 275, 9131–9135. [Google Scholar] [CrossRef] [Green Version]
- Ribon, V.; Johnson, J.H.; Camp, H.S.; Saltiel, A.R. Thiazolidinediones and insulin resistance: Peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc. Natl. Acad. Sci. USA 1998, 95, 14751–14756. [Google Scholar] [CrossRef]
- Liu, J.; DeYoung, S.M.; Hwang, J.B.; O’Leary, E.E.; Saltiel, A.R. The roles of Cbl-b and c-Cbl in insulin-stimulated glucose transport. J. Biol. Chem. 2003, 278, 36754–36762. [Google Scholar] [CrossRef] [Green Version]
- Kramer, D.; Shapiro, R.; Adler, A.; Bush, E.; Rondinone, C.M. Insulin-sensitizing effect of rosiglitazone (BRL-49653) by regulation of glucose transporters in muscle and fat of Zucker rats. Metabolism 2001, 50, 1294–1300. [Google Scholar] [CrossRef]
- Standaert, M.L.; Kanoh, Y.; Sajan, M.P.; Bandyopadhyay, G.; Farese, R.V. Cbl, IRS-1, and IRS-2 mediate effects of rosiglitazone on PI3K, PKC-lambda, and glucose transport in 3T3/L1 adipocytes. Endocrinology 2002, 143, 1705–1716. [Google Scholar] [CrossRef]
- Kim, H.I.; Kim, J.W.; Kim, S.H.; Cha, J.Y.; Kim, K.S.; Ahn, Y.H. Identification and functional characterization of the peroxisomal proliferator response element in rat GLUT2 promoter. Diabetes 2000, 49, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Samaha, F.F.; Szapary, P.O.; Iqbal, N.; Williams, M.M.; Bloedon, L.T.; Kochar, A.; Wolfe, M.L.; Rader, D.J. Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high-density lipoprotein cholesterol and metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C. Signalling links in the liver: Knitting SOCS with fat and inflammation. J. Hepatol. 2005, 43, 193–196. [Google Scholar] [CrossRef]
- Szalkowski, D.; White-Carrington, S.; Berger, J.; Zhang, B. Antidiabetic thiazolidinediones block the inhibitory effect of tumor necrosis factor-alpha on differentiation, insulin-stimulated glucose uptake, and gene expression in 3T3-L1 cells. Endocrinology 1995, 136, 1474–1481. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Hawkins, M.; Combs, T.P.; Rajala, M.W.; Doebber, T.; Berger, J.P.; Wagner, J.A.; Wu, M.; Knopps, A.; Xiang, A.H.; et al. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J. Biol. Chem. 2004, 279, 12152–12162. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Armoni, M.; Harel, C.; Karni, S.; Chen, H.; Bar-Yoseph, F.; Ver, M.R.; Quon, M.J.; Karnieli, E. FOXO1 represses peroxisome proliferator-activated receptor-gamma1 and -gamma2 gene promoters in primary adipocytes. A novel paradigm to increase insulin sensitivity. J. Biol. Chem. 2006, 281, 19881–19891. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Imamura, T.; Sonoda, N.; Sears, D.D.; Patsouris, D.; Kim, J.J.; Olefsky, J.M. FOXO1 transrepresses peroxisome proliferator-activated receptor gamma transactivation, coordinating an insulin-induced feed-forward response in adipocytes. J. Biol. Chem. 2009, 284, 12188–12197. [Google Scholar] [CrossRef] [Green Version]
- Dowell, P.; Otto, T.C.; Adi, S.; Lane, M.D. Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J. Biol. Chem. 2003, 278, 45485–45491. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.J.; Ludvik, B.; Beerdsen, P.; Joyce, M.; Olefsky, J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 1994, 331, 1188–1193. [Google Scholar] [CrossRef]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes 2003, 52, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.K.; Garg, A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J. Clin. Endocrinol. Metab. 2002, 87, 408–411. [Google Scholar] [CrossRef]
- Hegele, R.A.; Cao, H.; Frankowski, C.; Mathews, S.T.; Leff, T. PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes 2002, 51, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Demir, T.; Onay, H.; Savage, D.B.; Temeloglu, E.; Uzum, A.K.; Kadioglu, P.; Altay, C.; Ozen, S.; Demir, L.; Cavdar, U.; et al. Familial partial lipodystrophy linked to a novel peroxisome proliferator activator receptor -gamma (PPARG) mutation, H449L: A comparison of people with this mutation and those with classic codon 482 Lamin A/C (LMNA) mutations. Diabet Med. 2016, 33, 1445–1450. [Google Scholar] [CrossRef] [Green Version]
- Ludtke, A.; Buettner, J.; Schmidt, H.H.; Worman, H.J. New PPARG mutation leads to lipodystrophy and loss of protein function that is partially restored by a synthetic ligand. J. Med. Genet. 2007, 44, e88. [Google Scholar] [CrossRef] [Green Version]
- Ludtke, A.; Buettner, J.; Wu, W.; Muchir, A.; Schroeter, A.; Zinn-Justin, S.; Spuler, S.; Schmidt, H.H.; Worman, H.J. Peroxisome proliferator-activated receptor-gamma C190S mutation causes partial lipodystrophy. J. Clin. Endocrinol. Metab. 2007, 92, 2248–2255. [Google Scholar] [CrossRef] [Green Version]
- Reitman, M.L.; Arioglu, E.; Gavrilova, O.; Taylor, S.I. Lipoatrophy revisited. Trends Endocrinol. Metab. 2000, 11, 410–416. [Google Scholar]
- Jones, J.R.; Barrick, C.; Kim, K.A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.C.; Brown-Borg, H.M. Peroxisome proliferator-activated receptor gamma coactivator 1 in caloric restriction and other models of longevity. J. Gerontol. Biol. Sci. Med. Sci. 2005, 60, 1494–1509. [Google Scholar] [CrossRef]
- Carvalho, C.R.; Brenelli, S.L.; Silva, A.C.; Nunes, A.L.; Velloso, L.A.; Saad, M.J. Effect of aging on insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of rats. Endocrinology 1996, 137, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, S.; Picard, F.; Vamecq, J.; Gelman, L.; Potier, N.; Zeyer, D.; Dubuquoy, L.; Bac, P.; Champy, M.F.; Plunket, K.D.; et al. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol. Cell 2001, 8, 737–747. [Google Scholar]
- Dunn, F.L.; Higgins, L.S.; Fredrickson, J.; De Paoli, A.M.; group, I.N.T.S. Selective modulation of PPARgamma activity can lower plasma glucose without typical thiazolidinedione side-effects in patients with Type 2 diabetes. J. Diabetes Complicat. 2011, 25, 151–158. [Google Scholar] [CrossRef]
- Beekmann, K.; Rubio, L.; de Haan, L.H.; Actis-Goretta, L.; van der Burg, B.; van Bladeren, P.J.; Rietjens, I.M. The effect of quercetin and kaempferol aglycones and glucuronides on peroxisome proliferator-activated receptor-gamma (PPAR-gamma). Food Funct. 2015, 6, 1098–1107. [Google Scholar] [CrossRef]
- Fang, X.K.; Gao, J.; Zhu, D.N. Kaempferol and quercetin isolated from Euonymus alatus improve glucose uptake of 3T3-L1 cells without adipogenesis activity. Life Sci. 2008, 82, 615–622. [Google Scholar] [CrossRef]
- Takahashi, H.; Hara, H.; Goto, T.; Kamakari, K.; Wataru, N.; Mohri, S.; Takahashi, N.; Suzuki, H.; Shibata, D.; Kawada, T. 13-Oxo-9(Z),11(E),15(Z)-octadecatrienoic acid activates peroxisome proliferator-activated receptor gamma in adipocytes. Lipids 2015, 50, 3–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, M.; Cai, W.; Yu, L.; Feng, L.; Zhang, L.; Zang, Q.; Wang, Y.; Wang, D.; Chen, H.; et al. Dietary component isorhamnetin is a PPARgamma antagonist and ameliorates metabolic disorders induced by diet or leptin deficiency. Sci. Rep. 2016, 6, 19288. [Google Scholar] [CrossRef]
- Yamauchi, T.; Waki, H.; Kamon, J.; Murakami, K.; Motojima, K.; Komeda, K.; Miki, H.; Kubota, N.; Terauchi, Y.; Tsuchida, A.; et al. Inhibition of RXR and PPARgamma ameliorates diet-induced obesity and type 2 diabetes. J. Clin. Investig. 2001, 108, 1001–1013. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Murakami, K.; Motojima, K.; Komeda, K.; Ide, T.; Kubota, N.; Terauchi, Y.; Tobe, K.; et al. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor gamma (PPARgamma) deficiency and PPARgamma agonist improve insulin resistance. J. Biol. Chem. 2001, 276, 41245–41254. [Google Scholar] [CrossRef] [Green Version]
- Heikkinen, S.; Argmann, C.; Feige, J.N.; Koutnikova, H.; Champy, M.F.; Dali-Youcef, N.; Schadt, E.E.; Laakso, M.; Auwerx, J. The Pro12Ala PPARgamma2 variant determines metabolism at the gene-environment interface. Cell Metab. 2009, 9, 88–98. [Google Scholar] [CrossRef]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef]
- Altshuler, D.; Hirschhorn, J.N.; Klannemark, M.; Lindgren, C.M.; Vohl, M.C.; Nemesh, J.; Lane, C.R.; Schaffner, S.F.; Bolk, S.; Brewer, C.; et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000, 26, 76–80. [Google Scholar] [CrossRef]
- Rieusset, J.; Touri, F.; Michalik, L.; Escher, P.; Desvergne, B.; Niesor, E.; Wahli, W. A new selective peroxisome proliferator-activated receptor gamma antagonist with antiobesity and antidiabetic activity. Mol. Endocrinol. 2002, 16, 2628–2644. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; Hoener, P.A.; Jow, L.; Bilakovics, J.; Klausing, K.; Mais, D.E.; Faulkner, A.; Croston, G.E.; Paterniti, J.R., Jr. A selective peroxisome proliferator-activated receptor-gamma (PPARgamma) modulator blocks adipocyte differentiation but stimulates glucose uptake in 3T3-L1 adipocytes. Mol. Endocrinol. 2000, 14, 1425–1433. [Google Scholar] [CrossRef]
- Ferrara, N.; Rinaldi, B.; Corbi, G.; Conti, V.; Stiuso, P.; Boccuti, S.; Rengo, G.; Rossi, F.; Filippelli, A. Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res. 2008, 11, 139–150. [Google Scholar] [CrossRef]
- Corbi, G.; Conti, V.; Scapagnini, G.; Filippelli, A.; Ferrara, N. Role of sirtuins, calorie restriction and physical activity in aging. Front. Biosci. (Elite Ed.) 2012, 4, 768–778. [Google Scholar]
- Rack, J.G.; Morra, R.; Barkauskaite, E.; Kraehenbuehl, R.; Ariza, A.; Qu, Y.; Ortmayer, M.; Leidecker, O.; Cameron, D.R.; Matic, I.; et al. Identification of a Class of Protein ADP-Ribosylating Sirtuins in Microbial Pathogens. Mol. Cell 2015, 59, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.G.; Tanny, J.C.; Kruger, R.G.; Walz, T.; Moazed, D. Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell 2005, 121, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef]
- Dang, W. The controversial world of sirtuins. Drug Discov. Today Technol. 2014, 12, e9–e17. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010, 12, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Preyat, N.; Leo, O. Sirtuin deacylases: A molecular link between metabolism and immunity. J. Leukoc. Biol. 2013, 93, 669–680. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Ben-Josef, G.; West, T.; Wozniak, D.F.; Holtzman, D.M.; Herzog, E.D.; Imai, S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J. Neurosci. 2010, 30, 10220–10232. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, R.; Faulkner, S.; Miyokawa, R.; Burhenn, L.; Henriksen, M.; Wood, J.G.; Helfand, S.L. Increased expression of Drosophila Sir2 extends life span in a dose-dependent manner. Aging (Albany N. Y.) 2013, 5, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.J.; Defossez, P.A.; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef] [Green Version]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef] [Green Version]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10042–10047. [Google Scholar] [CrossRef] [Green Version]
- van der Horst, A.; Tertoolen, L.G.; de Vries-Smits, L.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [Green Version]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Gross, D.N.; van den Heuvel, A.P.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef] [Green Version]
- Frescas, D.; Valenti, L.; Accili, D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005, 280, 20589–20595. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006, 4, e31. [Google Scholar] [CrossRef]
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Meneur, C.; Permutt, M.A.; Imai, S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005, 2, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Bae, N.S.; Swanson, M.J.; Vassilev, A.; Howard, B.H. Human histone deacetylase SIRT2 interacts with the homeobox transcription factor HOXA10. J. Biochem. 2004, 135, 695–700. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.S.; Tainsky, M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell Biol. 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Starai, V.J.; Celic, I.; Cole, R.N.; Boeke, J.D.; Escalante-Semerena, J.C. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 2002, 298, 2390–2392. [Google Scholar] [CrossRef]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [Green Version]
- Rose, G.; Dato, S.; Altomare, K.; Bellizzi, D.; Garasto, S.; Greco, V.; Passarino, G.; Feraco, E.; Mari, V.; Barbi, C.; et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 2003, 38, 1065–1070. [Google Scholar] [CrossRef]
- Nasrin, N.; Wu, X.; Fortier, E.; Feng, Y.; Bare, O.C.; Chen, S.; Ren, X.; Wu, Z.; Streeper, R.S.; Bordone, L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 2010, 285, 31995–32002. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- McCord, R.A.; Michishita, E.; Hong, T.; Berber, E.; Boxer, L.D.; Kusumoto, R.; Guan, S.; Shi, X.; Gozani, O.; Burlingame, A.L.; et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany N. Y.) 2009, 1, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Luo, L.L.; Chen, X.C.; Fu, Y.C.; Xu, J.J.; Li, L.; Lin, X.H.; Xiang, Y.F.; Zhang, X.M. The effects of caloric restriction and a high-fat diet on ovarian lifespan and the expression of SIRT1 and SIRT6 proteins in rats. Aging Clin. Exp. Res. 2012, 24, 125–133. [Google Scholar] [CrossRef]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef]
- Geng, Y.Q.; Li, T.T.; Liu, X.Y.; Li, Z.H.; Fu, Y.C. SIRT1 and SIRT5 activity expression and behavioral responses to calorie restriction. J. Cell Biochem. 2011, 112, 3755–3761. [Google Scholar] [CrossRef]
- Ran, M.; Li, Z.; Yang, L.; Tong, L.; Zhang, L.; Dong, H. Calorie restriction attenuates cerebral ischemic injury via increasing SIRT1 synthesis in the rat. Brain Res. 2015, 1610, 61–68. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Yu, W.; Zhou, H.F.; Lin, R.B.; Fu, Y.C.; Wang, W. Shortterm calorie restriction activates SIRT14 and 7 in cardiomyocytes in vivo and in vitro. Mol. Med. Rep. 2014, 9, 1218–1224. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Qin, J.; Chen, C.; Fu, Y.; Wang, W. Moderate calorie restriction attenuates ageassociated alterations and improves cardiac function by increasing SIRT1 and SIRT3 expression. Mol. Med. Rep. 2018, 18, 4087–4094. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Deng, X.Q.; Li, N.X. Effects of calorie restriction on SIRT1 expression in liver of nonalcoholic fatty liver disease: Experiment with rats. Zhonghua Yi Xue Za Zhi 2007, 87, 1434–1437. [Google Scholar]
- Boily, G.; Seifert, E.L.; Bevilacqua, L.; He, X.H.; Sabourin, G.; Estey, C.; Moffat, C.; Crawford, S.; Saliba, S.; Jardine, K.; et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE 2008, 3, e1759. [Google Scholar] [CrossRef]
- Bordone, L.; Cohen, D.; Robinson, A.; Motta, M.C.; van Veen, E.; Czopik, A.; Steele, A.D.; Crowe, H.; Marmor, S.; Luo, J.; et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 2007, 6, 759–767. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Hubbard, B.P. Lifespan and healthspan extension by resveratrol. Biochim. Biophys. Acta 2015, 1852, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Barger, J.L.; Kayo, T.; Vann, J.M.; Arias, E.B.; Wang, J.; Hacker, T.A.; Wang, Y.; Raederstorff, D.; Morrow, J.D.; Leeuwenburgh, C.; et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE 2008, 3, e2264. [Google Scholar] [CrossRef]
- Laurent, G.; de Boer, V.C.; Finley, L.W.; Sweeney, M.; Lu, H.; Schug, T.T.; Cen, Y.; Jeong, S.M.; Li, X.; Sauve, A.A.; et al. SIRT4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Mol. Cell Biol. 2013, 33, 4552–4561. [Google Scholar] [CrossRef] [Green Version]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Zhai, P.; Yamamoto, T.; Ikeda, Y.; Byun, J.; Hsu, C.P.; Sadoshima, J. Peroxisome Proliferator Activated Receptor-alpha Association With Silent Information Regulator 1 Suppresses Cardiac Fatty Acid Metabolism in the Failing Heart. Circ. Heart Fail. 2015, 8, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Alcendor, R.; Zhai, P.; Park, J.Y.; Shao, D.; Cho, J.; Yamamoto, T.; Tian, B.; Sadoshima, J. PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011, 14, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Zhai, P.; Alcendor, R.; Park, J.Y.; Tian, B.; Sadoshima, J. Suppression of ERR targets by a PPARalpha/Sirt1 complex in the failing heart. Cell Cycle 2012, 11, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Iwasaki, Y.; Nishiyama, M.; Taguchi, T.; Tsugita, M.; Nakayama, S.; Kambayashi, M.; Hashimoto, K.; Terada, Y. PPARbeta/delta regulates the human SIRT1 gene transcription via Sp1. Endocr. J. 2010, 57, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Barroso, E.; Eyre, E.; Palomer, X.; Vazquez-Carrera, M. The peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) agonist GW501516 prevents TNF-alpha-induced NF-kappaB activation in human HaCaT cells by reducing p65 acetylation through AMPK and SIRT1. Biochem. Pharmacol. 2011, 81, 534–543. [Google Scholar] [CrossRef]
- Gong, K.; Qu, B.; Wang, C.; Zhou, J.; Liao, D.; Zheng, W.; Pan, X. Peroxisome Proliferator-Activated Receptor alpha Facilitates Osteogenic Differentiation in MC3T3-E1 Cells via the Sirtuin 1-Dependent Signaling Pathway. Mol. Cells 2017, 40, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Gong, K.; Qu, B.; Liao, D.; Liu, D.; Wang, C.; Zhou, J.; Pan, X. MiR-132 regulates osteogenic differentiation via downregulating Sirtuin1 in a peroxisome proliferator-activated receptor beta/delta-dependent manner. Biochem. Biophys. Res. Commun. 2016, 478, 260–267. [Google Scholar] [CrossRef]
- Qu, B.; Ma, Y.; Yan, M.; Gong, K.; Liang, F.; Deng, S.; Jiang, K.; Ma, Z.; Pan, X. Sirtuin1 promotes osteogenic differentiation through downregulation of peroxisome proliferator-activated receptor gamma in MC3T3-E1 cells. Biochem. Biophys. Res. Commun. 2016, 478, 439–445. [Google Scholar] [CrossRef]
- Kim, M.Y.; Kang, E.S.; Ham, S.A.; Hwang, J.S.; Yoo, T.S.; Lee, H.; Paek, K.S.; Park, C.; Lee, H.T.; Kim, J.H.; et al. The PPARdelta-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem. Pharmacol. 2012, 84, 1627–1634. [Google Scholar] [CrossRef]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Y.; Xiao, F.; Liu, Y.; Wang, J.; Gao, H.; Rong, S.; Yao, Y.; Li, J.; Xu, G. The peroxisome proliferator-activated receptor gamma agonist pioglitazone prevents NF-kappaB activation in cisplatin nephrotoxicity through the reduction of p65 acetylation via the AMPK-SIRT1/p300 pathway. Biochem. Pharmacol. 2016, 101, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Floyd, Z.E.; Wang, Z.Q.; Kilroy, G.; Cefalu, W.T. Modulation of peroxisome proliferator-activated receptor gamma stability and transcriptional activity in adipocytes by resveratrol. Metabolism 2008, 57, S32–S38. [Google Scholar] [CrossRef] [Green Version]
- Hagen, T.M. Oxidative stress, redox imbalance, and the aging process. Antioxid. Redox Signal. 2003, 5, 503–506. [Google Scholar] [CrossRef]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 2007, 292, C670–C686. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Merry, B.J. Molecular mechanisms linking calorie restriction and longevity. Int. J. Biochem. Cell Biol. 2002, 34, 1340–1354. [Google Scholar]
- Sanz, A.; Caro, P.; Ibanez, J.; Gomez, J.; Gredilla, R.; Barja, G. Dietary restriction at old age lowers mitochondrial oxygen radical production and leak at complex I and oxidative DNA damage in rat brain. J. Bioenerg. Biomembr. 2005, 37, 83–90. [Google Scholar] [CrossRef]
- Yu, B.P. Aging and oxidative stress: Modulation by dietary restriction. Free Radic. Biol. Med. 1996, 21, 651–668. [Google Scholar] [CrossRef]
- Barja, G. Aging in vertebrates, and the effect of caloric restriction: A mitochondrial free radical production-DNA damage mechanism? Biol. Rev. Cambr. Philos. Soc. 2004, 79, 235–251. [Google Scholar]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Forster, M.J.; Sohal, B.H.; Sohal, R.S. Reversible effects of long-term caloric restriction on protein oxidative damage. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, B522–B529. [Google Scholar]
- Lambert, A.J.; Merry, B.J. Lack of effect of caloric restriction on bioenergetics and reactive oxygen species production in intact rat hepatocytes. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 175–180. [Google Scholar]
- Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Merry, B.J. Effect of ageing and caloric restriction on specific markers of protein oxidative damage and membrane peroxidizability in rat liver mitochondria. Mech. Ageing Dev. 2004, 125, 529–538. [Google Scholar] [CrossRef]
- Bagi, Z.; Koller, A.; Kaley, G. PPARgamma activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H742–H748. [Google Scholar] [CrossRef] [Green Version]
- Meerarani, P.; Reiterer, G.; Toborek, M.; Hennig, B. Zinc modulates PPARgamma signaling and activation of porcine endothelial cells. J. Nutr. 2003, 133, 3058–3064. [Google Scholar] [CrossRef] [Green Version]
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Tanabe, T.; Takanashi, M.; Irukayama-Tomobe, Y.; Sakai, S.; Ohmori, H.; Matsuda, M.; Yamaguchi, I. Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1750–H1760. [Google Scholar] [CrossRef]
- Patterson, A.D.; Shah, Y.M.; Matsubara, T.; Krausz, K.W.; Gonzalez, F.J. Peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 protects against acetaminophen-induced liver toxicity. Hepatology 2012, 56, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Sekulic-Jablanovic, M.; Petkovic, V.; Wright, M.B.; Kucharava, K.; Huerzeler, N.; Levano, S.; Brand, Y.; Leitmeyer, K.; Glutz, A.; Bausch, A.; et al. Effects of peroxisome proliferator activated receptors (PPAR)-gamma and -alpha agonists on cochlear protection from oxidative stress. PLoS ONE 2017, 12, e0188596. [Google Scholar] [CrossRef]
- Girnun, G.D.; Domann, F.E.; Moore, S.A.; Robbins, M.E. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 2002, 16, 2793–2801. [Google Scholar] [CrossRef] [Green Version]
- Okuno, Y.; Matsuda, M.; Miyata, Y.; Fukuhara, A.; Komuro, R.; Shimabukuro, M.; Shimomura, I. Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a response element distinct from that of mouse. Endocr. J. 2010, 57, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Fu, M.; Qin, Q.; Lewis, W.; Kim, H.W.; Fukai, T.; Bacanamwo, M.; Chen, Y.E.; Schneider, M.D.; Mangelsdorf, D.J.; et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc. Res. 2007, 76, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.S.; Kim, M.; Youn, B.S.; Lee, N.S.; Park, J.W.; Lee, I.K.; Lee, Y.S.; Kim, J.B.; Cho, Y.M.; Lee, H.K.; et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol. Cell Biol. 2009, 29, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Liu, F.; Yan, M.; Ji, H.; Hu, L.; Li, X.; Qian, J.; He, X.; Zhang, L.; Shen, A.; et al. Peroxisome proliferator-activated receptor-gamma agonists suppress iNOS expression induced by LPS in rat primary Schwann cells. J. Neuroimmunol. 2010, 218, 36–47. [Google Scholar] [CrossRef]
- Vandewalle, B.; Moerman, E.; Lefebvre, B.; Defrance, F.; Gmyr, V.; Lukowiak, B.; Kerr Conte, J.; Pattou, F. PPARgamma-dependent and -independent effects of rosiglitazone on lipotoxic human pancreatic islets. Biochem. Biophys. Res. Commun. 2008, 366, 1096–1101. [Google Scholar] [CrossRef]
- Kleinhenz, J.M.; Kleinhenz, D.J.; You, S.; Ritzenthaler, J.D.; Hansen, J.M.; Archer, D.R.; Sutliff, R.L.; Hart, C.M. Disruption of endothelial peroxisome proliferator-activated receptor-gamma reduces vascular nitric oxide production. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1647–H1654. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Li, M.; Pascual, G.; Glass, C.K. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol. Cell Biol. 2000, 20, 4699–4707. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Chaki, R.; Mandal, V.; Mandal, S.C. COX-2 as a target for cancer chemotherapy. Pharmacol. Rep. 2010, 62, 233–244. [Google Scholar]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Z.; Liu, J.Z.; Hu, J.X.; Chen, H.L.; Li, W.L.; Hai, C.X. Double antioxidant activities of rosiglitazone against high glucose-induced oxidative stress in hepatocyte. Toxicol. In Vitro 2011, 25, 839–847. [Google Scholar] [CrossRef]
- Aleshin, S.; Grabeklis, S.; Hanck, T.; Sergeeva, M.; Reiser, G. Peroxisome proliferator-activated receptor (PPAR)-gamma positively controls and PPARalpha negatively controls cyclooxygenase-2 expression in rat brain astrocytes through a convergence on PPARbeta/delta via mutual control of PPAR expression levels. Mol. Pharmacol. 2009, 76, 414–424. [Google Scholar] [CrossRef]
- Collino, M.; Aragno, M.; Mastrocola, R.; Gallicchio, M.; Rosa, A.C.; Dianzani, C.; Danni, O.; Thiemermann, C.; Fantozzi, R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur. J. Pharmacol. 2006, 530, 70–80. [Google Scholar] [CrossRef]
- Zhao, Y.; Patzer, A.; Herdegen, T.; Gohlke, P.; Culman, J. Activation of cerebral peroxisome proliferator-activated receptors gamma promotes neuroprotection by attenuation of neuronal cyclooxygenase-2 overexpression after focal cerebral ischemia in rats. FASEB J. 2006, 20, 1162–1175. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.H.; Wu, K.L.; Kung, P.S.; Chan, J.Y. Oral intake of rosiglitazone promotes a central antihypertensive effect via upregulation of peroxisome proliferator-activated receptor-gamma and alleviation of oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertension 2010, 55, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Clavel, S.; Bouillaud, F.; Ricquier, D.; Richard, D. Uncoupling protein 2: A novel player in neuroprotection. Trends Mol. Med. 2003, 9, 522–525. [Google Scholar]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef]
- Fuenzalida, K.; Quintanilla, R.; Ramos, P.; Piderit, D.; Fuentealba, R.A.; Martinez, G.; Inestrosa, N.C.; Bronfman, M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 2007, 282, 37006–37015. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Sun, C.; Sun, Y.; Tan, H.; Wu, Y.; Cui, B.; Wu, Z. PPAR gamma protects cardiomyocytes against oxidative stress and apoptosis via Bcl-2 upregulation. Vasc. Pharmacol. 2009, 51, 169–174. [Google Scholar] [CrossRef]
- Chung, S.W.; Kang, B.Y.; Kim, S.H.; Pak, Y.K.; Cho, D.; Trinchieri, G.; Kim, T.S. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J. Biol. Chem. 2000, 275, 32681–32687. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xia, Q.; Zhang, Q.; Li, H.; Zhang, J.; Li, A.; Xiu, R. Peroxisome proliferator-activated receptor-gamma ligands 15-deoxy-delta(12,14)-prostaglandin J2 and pioglitazone inhibit hydroxyl peroxide-induced TNF-alpha and lipopolysaccharide-induced CXC chemokine expression in neonatal rat cardiac myocytes. Shock 2009, 32, 317–324. [Google Scholar] [CrossRef]
- Goto, M.; Katayama, K.I.; Shirakawa, F.; Tanaka, I. Involvement of NF-kappaB p50/p65 heterodimer in activation of the human pro-interleukin-1beta gene at two subregions of the upstream enhancer element. Cytokine 1999, 11, 16–28. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, K.W.; Yu, B.P.; Chung, H.Y. The effect of age on cyclooxygenase-2 gene expression: NF-kappaB activation and IkappaBalpha degradation. Free Radic. Biol. Med. 2000, 28, 683–692. [Google Scholar] [CrossRef]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef]
- Yao, J.; Mackman, N.; Edgington, T.S.; Fan, S.T. Lipopolysaccharide induction of the tumor necrosis factor-alpha promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-kappaB transcription factors. J. Biol. Chem. 1997, 272, 17795–17801. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; de Vera, M.E.; Ganster, R.W.; Wang, Q.; Shapiro, R.A.; Morris, S.M., Jr.; Billiar, T.R.; Geller, D.A. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J. Biol. Chem. 1998, 273, 15148–15156. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-kappaB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243. [Google Scholar] [CrossRef]
- Ye, J. Regulation of PPARgamma function by TNF-alpha. Biochem. Biophys. Res. Commun. 2008, 374, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, M.; Costa, L.; Palermo, R.; Giovenco, A.; Vacca, A.; Gulino, A. Protective effect of pioglitazone, a PPARgamma ligand, in a 3 nitropropionic acid model of Huntington’s disease. Brain Res. Bull. 2011, 85, 231–237. [Google Scholar] [CrossRef]
- Osburn, W.O.; Kensler, T.W. Nrf2 signaling: An adaptive response pathway for protection against environmental toxic insults. Mutat. Res. 2008, 659, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Cho, I.J.; Kim, S.G. Transactivation of the PPAR-responsive enhancer module in chemopreventive glutathione S-transferase gene by the peroxisome proliferator-activated receptor-gamma and retinoid X receptor heterodimer. Cancer Res. 2004, 64, 3701–3713. [Google Scholar] [CrossRef] [Green Version]
- D’Archivio, M.; Scazzocchio, B.; Filesi, C.; Vari, R.; Maggiorella, M.T.; Sernicola, L.; Santangelo, C.; Giovannini, C.; Masella, R. Oxidised LDL up-regulate CD36 expression by the Nrf2 pathway in 3T3-L1 preadipocytes. FEBS Lett. 2008, 582, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Gong, P.; Stewart, D.; Hu, B.; Li, N.; Cook, J.; Nel, A.; Alam, J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Delta(12,14)-prostaglandin J(2) is mediated by the stress response elements and transcription factor Nrf2. Antioxid. Redox Signal. 2002, 4, 249–257. [Google Scholar] [CrossRef]
- Nakaso, K.; Yano, H.; Fukuhara, Y.; Takeshima, T.; Wada-Isoe, K.; Nakashima, K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003, 546, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Tabbi-Anneni, I.; Gunda, V.; Wang, L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1211–G1221. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R18–R36. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.C.; Warshaw, J.B.; Sanadi, D.R. Regulation of mitochondrial respiration in senescence. J. Cell Physiol. 1972, 80, 141–148. [Google Scholar] [CrossRef]
- Hansford, R.G. Lipid oxidation by heart mitochondria from young adult and senescent rats. Biochem. J. 1978, 170, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Trounce, I.; Byrne, E.; Marzuki, S. Decline in skeletal muscle mitochondrial respiratory chain function: Possible factor in ageing. Lancet 1989, 1, 637–639. [Google Scholar]
- Cooper, J.M.; Mann, V.M.; Schapira, A.H. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: Effect of ageing. J. Neurol. Sci. 1992, 113, 91–98. [Google Scholar]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Solarino, G.; Santacroce, G.; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 1, 642–645. [Google Scholar]
- Hancock, C.R.; Han, D.H.; Higashida, K.; Kim, S.H.; Holloszy, J.O. Does calorie restriction induce mitochondrial biogenesis? A reevaluation. FASEB J. 2011, 25, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; de Cabo, R.; Lane, M.A.; Ingram, D.K. Caloric restriction modulates early events in insulin signaling in liver and skeletal muscle of rat. Ann. N. Y. Acad. Sci. 2004, 1019, 448–452. [Google Scholar] [CrossRef]
- Ranhotra, H.S. Long-term caloric restriction up-regulates PPAR gamma co-activator 1 alpha (PGC-1alpha) expression in mice. Indian J. Biochem. Biophys. 2010, 47, 272–277. [Google Scholar]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Lanza, I.R.; Zabielski, P.; Klaus, K.A.; Morse, D.M.; Heppelmann, C.J.; Bergen, H.R., 3rd; Dasari, S.; Walrand, S.; Short, K.R.; Johnson, M.L.; et al. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 2012, 16, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Hepple, R.T.; Baker, D.J.; McConkey, M.; Murynka, T.; Norris, R. Caloric restriction protects mitochondrial function with aging in skeletal and cardiac muscles. Rejuvenation Res. 2006, 9, 219–222. [Google Scholar] [CrossRef]
- Miller, B.F.; Robinson, M.M.; Bruss, M.D.; Hellerstein, M.; Hamilton, K.L. A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 2012, 11, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [Green Version]
- Gudiksen, A.; Pilegaard, H. PGC-1alpha and fasting-induced PDH regulation in mouse skeletal muscle. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Geng, T.; Li, P.; Okutsu, M.; Yin, X.; Kwek, J.; Zhang, M.; Yan, Z. PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 2010, 298, C572–C579. [Google Scholar] [CrossRef] [Green Version]
- Leick, L.; Wojtaszewski, J.F.; Johansen, S.T.; Kiilerich, K.; Comes, G.; Hellsten, Y.; Hidalgo, J.; Pilegaard, H. PGC-1alpha is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E463–E474. [Google Scholar] [CrossRef] [Green Version]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [Green Version]
- Perez-Schindler, J.; Summermatter, S.; Salatino, S.; Zorzato, F.; Beer, M.; Balwierz, P.J.; van Nimwegen, E.; Feige, J.N.; Auwerx, J.; Handschin, C. The corepressor NCoR1 antagonizes PGC-1alpha and estrogen-related receptor alpha in the regulation of skeletal muscle function and oxidative metabolism. Mol. Cell Biol. 2012, 32, 4913–4924. [Google Scholar] [CrossRef] [Green Version]
- Zolezzi, J.M.; Silva-Alvarez, C.; Ordenes, D.; Godoy, J.A.; Carvajal, F.J.; Santos, M.J.; Inestrosa, N.C. Peroxisome proliferator-activated receptor (PPAR) gamma and PPARalpha agonists modulate mitochondrial fusion-fission dynamics: Relevance to reactive oxygen species (ROS)-related neurodegenerative disorders? PLoS ONE 2013, 8, e64019. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Fujii, H.; Takahashi, T.; Kodama, M.; Aizawa, Y.; Ohta, Y.; Ono, T.; Hasegawa, G.; Naito, M.; Nakajima, T.; et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J. Biol. Chem. 2000, 275, 22293–22299. [Google Scholar] [CrossRef] [Green Version]
- Minnich, A.; Tian, N.; Byan, L.; Bilder, G. A potent PPARalpha agonist stimulates mitochondrial fatty acid beta-oxidation in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E270–E279. [Google Scholar] [CrossRef] [Green Version]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M.; Barth, E.; Cagnard, N.; Montagner, A.; Chretien, D.; Rugarli, E.I.; Guillou, H.; et al. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARalpha. Nat. Commun. 2019, 10, 1566. [Google Scholar] [CrossRef] [Green Version]
- Cree, M.G.; Zwetsloot, J.J.; Herndon, D.N.; Qian, T.; Morio, B.; Fram, R.; Sanford, A.P.; Aarsland, A.; Wolfe, R.R. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann. Surg. 2007, 245, 214–221. [Google Scholar] [CrossRef]
- Hong, M.; Song, K.D.; Lee, H.K.; Yi, S.; Lee, Y.S.; Heo, T.H.; Jun, H.S.; Kim, S.J. Fibrates inhibit the apoptosis of Batten disease lymphoblast cells via autophagy recovery and regulation of mitochondrial membrane potential. In Vitro Cell Dev. Biol. Anim. 2016, 52, 349–355. [Google Scholar] [CrossRef]
- Mohagheghi, F.; Ahmadiani, A.; Rahmani, B.; Moradi, F.; Romond, N.; Khalaj, L. Gemfibrozil pretreatment resulted in a sexually dimorphic outcome in the rat models of global cerebral ischemia-reperfusion via modulation of mitochondrial pro-survival and apoptotic cell death factors as well as MAPKs. J. Mol. Neurosci. 2013, 50, 379–393. [Google Scholar] [CrossRef]
- Brunmair, B.; Lest, A.; Staniek, K.; Gras, F.; Scharf, N.; Roden, M.; Nohl, H.; Waldhausl, W.; Furnsinn, C. Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complex I. J. Pharmacol. Exp. Ther. 2004, 311, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wallace, K.B. The effect of peroxisome proliferators on mitochondrial bioenergetics. Toxicol. Sci. 1999, 48, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Zungu, M.; Young, M.E.; Stanley, W.C.; Essop, M.F. Chronic treatment with the peroxisome proliferator-activated receptor alpha agonist Wy-14,643 attenuates myocardial respiratory capacity and contractile function. Mol. Cell Biochem. 2009, 330, 55–62. [Google Scholar] [CrossRef]
- Koh, J.H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.H. PPARbeta Is Essential for Maintaining Normal Levels of PGC-1alpha and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab. 2017, 25, 1176–1185. [Google Scholar] [CrossRef]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef] [Green Version]
- Dulloo, A.G.; Samec, S. Uncoupling proteins: Their roles in adaptive thermogenesis and substrate metabolism reconsidered. Br. J. Nutr. 2001, 86, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Kleiner, S.; Nguyen-Tran, V.; Bare, O.; Huang, X.; Spiegelman, B.; Wu, Z. PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function. J. Biol. Chem. 2009, 284, 18624–18633. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARdelta Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, J.; Huang, J.; Li, T.; Xu, D.; Zuo, B.; Hou, L.; Wu, W.; Zhang, L.; Xia, X.; et al. The formation of brown adipose tissue induced by transgenic over-expression of PPARgamma2. Biochem. Biophys. Res. Commun. 2014, 446, 959–964. [Google Scholar] [CrossRef]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Investig. 2004, 114, 1281–1289. [Google Scholar] [CrossRef]
- Hakansson, J.; Eliasson, B.; Smith, U.; Enerback, S. Adipocyte mitochondrial genes and the forkhead factor FOXC2 are decreased in type 2 diabetes patients and normalized in response to rosiglitazone. Diabetol. Metab. Syndr. 2011, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Rong, J.X.; Klein, J.L.; Qiu, Y.; Xie, M.; Johnson, J.H.; Waters, K.M.; Zhang, V.; Kashatus, J.A.; Remlinger, K.S.; Bing, N.; et al. Rosiglitazone Induces Mitochondrial Biogenesis in Differentiated Murine 3T3-L1 and C3H/10T1/2 Adipocytes. PPAR Res. 2011, 2011, 179454. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; Frauwirth, K.A.; Rangwala, S.M.; Lazar, M.A.; Thompson, C.B. Thiazolidinedione activation of peroxisome proliferator-activated receptor gamma can enhance mitochondrial potential and promote cell survival. J. Biol. Chem. 2002, 277, 31781–31788. [Google Scholar] [CrossRef] [Green Version]
- Chiang, M.C.; Chern, Y.; Huang, R.N. PPARgamma rescue of the mitochondrial dysfunction in Huntington’s disease. Neurobiol. Dis. 2012, 45, 322–328. [Google Scholar] [CrossRef]
- Dello Russo, C.; Gavrilyuk, V.; Weinberg, G.; Almeida, A.; Bolanos, J.P.; Palmer, J.; Pelligrino, D.; Galea, E.; Feinstein, D.L. Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J. Biol. Chem. 2003, 278, 5828–5836. [Google Scholar] [CrossRef] [Green Version]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef]
- Ghosh, S.; Patel, N.; Rahn, D.; McAllister, J.; Sadeghi, S.; Horwitz, G.; Berry, D.; Wang, K.X.; Swerdlow, R.H. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol. Pharmacol. 2007, 71, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, D.L.; Spagnolo, A.; Akar, C.; Weinberg, G.; Murphy, P.; Gavrilyuk, V.; Dello Russo, C. Receptor-independent actions of PPAR thiazolidinedione agonists: Is mitochondrial function the key? Biochem. Pharmacol. 2005, 70, 177–188. [Google Scholar] [CrossRef]
- Colca, J.R.; Tanis, S.P.; McDonald, W.G.; Kletzien, R.F. Insulin sensitizers in 2013: New insights for the development of novel therapeutic agents to treat metabolic diseases. Expert Opin. Investig. Drugs 2014, 23, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, M.V.; Weigle, W.O.; Noonan, D.J.; Torbett, B.E.; McEvilly, R.J.; Koch, R.J.; Cardenas, G.J.; Ernst, D.N. Patterns of cytokine gene expression by CD4+ T cells from young and old mice. J. Immunol. 1993, 150, 3602–3614. [Google Scholar]
- Riancho, J.A.; Zarrabeitia, M.T.; Amado, J.A.; Olmos, J.M.; Gonzalez-Macias, J. Age-related differences in cytokine secretion. Gerontology 1994, 40, 8–12. [Google Scholar] [CrossRef]
- Miller, R.A. Aging and immune function. Int. Rev. Cytol. 1991, 124, 187–215. [Google Scholar] [CrossRef]
- Kubo, M.; Cinader, B. Polymorphism of age-related changes in interleukin (IL) production: Differential changes of T helper subpopulations, synthesizing IL 2, IL 3 and IL 4. Eur. J. Immunol. 1990, 20, 1289–1296. [Google Scholar] [CrossRef]
- Hayek, M.G.; Mura, C.; Wu, D.; Beharka, A.A.; Han, S.N.; Paulson, K.E.; Hwang, D.; Meydani, S.N. Enhanced expression of inducible cyclooxygenase with age in murine macrophages. J. Immunol. 1997, 159, 2445–2451. [Google Scholar]
- Erol, A. Interleukin-6 (IL-6) is still the leading biomarker of the metabolic and aging related disorders. Med. Hypotheses 2007, 69, 708. [Google Scholar] [CrossRef]
- Ricote, M.; Huang, J.; Fajas, L.; Li, A.; Welch, J.; Najib, J.; Witztum, J.L.; Auwerx, J.; Palinski, W.; Glass, C.K. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 7614–7619. [Google Scholar]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Okada, M.; Yan, S.F.; Pinsky, D.J. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB J. 2002, 16, 1861–1868. [Google Scholar] [CrossRef]
- Barbier, O.; Torra, I.P.; Duguay, Y.; Blanquart, C.; Fruchart, J.C.; Glineur, C.; Staels, B. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 717–726. [Google Scholar]
- Narala, V.R.; Adapala, R.K.; Suresh, M.V.; Brock, T.G.; Peters-Golden, M.; Reddy, R.C. Leukotriene B4 is a physiologically relevant endogenous peroxisome proliferator-activated receptor-alpha agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef] [Green Version]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar]
- Crisafulli, C.; Cuzzocrea, S. The role of endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha (PPAR-alpha) in the regulation of inflammation in macrophages. Shock 2009, 32, 62–73. [Google Scholar] [CrossRef]
- Peters, J.M.; Hollingshead, H.E.; Gonzalez, F.J. Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin. Sci. (Lond.) 2008, 115, 107–127. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Cheng, L.; Qin, Q.; Frontin, S.; Yang, Q. PPARdelta modulates lipopolysaccharide-induced TNFalpha inflammation signaling in cultured cardiomyocytes. J. Mol. Cell Cardiol. 2006, 40, 821–828. [Google Scholar] [CrossRef]
- Iwashita, A.; Muramatsu, Y.; Yamazaki, T.; Muramoto, M.; Kita, Y.; Yamazaki, S.; Mihara, K.; Moriguchi, A.; Matsuoka, N. Neuroprotective efficacy of the peroxisome proliferator-activated receptor delta-selective agonists in vitro and in vivo. J. Pharmacol. Exp. Ther. 2007, 320, 1087–1096. [Google Scholar] [CrossRef]
- Benetti, E.; Mastrocola, R.; Rogazzo, M.; Chiazza, F.; Aragno, M.; Fantozzi, R.; Collino, M.; Minetto, M.A. High sugar intake and development of skeletal muscle insulin resistance and inflammation in mice: A protective role for PPAR- delta agonism. Mediat. Inflamm. 2013, 2013, 509502. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Lichtenstein, G.R.; Stein, R.B.; Deren, J.J.; Judge, T.A.; Fogt, F.; Furth, E.E.; Demissie, E.J.; Hurd, L.B.; Su, C.G.; et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis. Am. J. Gastroenterol. 2001, 96, 3323–3328. [Google Scholar] [CrossRef]
- Liang, H.L.; Ouyang, Q. A clinical trial of combined use of rosiglitazone and 5-aminosalicylate for ulcerative colitis. World J. Gastroenterol. WJG 2008, 14, 114–119. [Google Scholar]
- Bassaganya-Riera, J.; Hontecillas, R. CLA and n-3 PUFA differentially modulate clinical activity and colonic PPAR-responsive gene expression in a pig model of experimental IBD. Clin. Nutr. 2006, 25, 454–465. [Google Scholar] [CrossRef]
- Hontecillas, R.; Wannemeulher, M.J.; Zimmerman, D.R.; Hutto, D.L.; Wilson, J.H.; Ahn, D.U.; Bassaganya-Riera, J. Nutritional regulation of porcine bacterial-induced colitis by conjugated linoleic acid. J. Nutr. 2002, 132, 2019–2027. [Google Scholar]
- Sanchez-Hidalgo, M.; Martin, A.R.; Villegas, I.; de la Lastra, C.A. Rosiglitazone, a PPARgamma ligand, modulates signal transduction pathways during the development of acute TNBS-induced colitis in rats. Eur. J. Pharmacol. 2007, 562, 247–258. [Google Scholar] [CrossRef]
- Sato, N.; Kozar, R.A.; Zou, L.; Weatherall, J.M.; Attuwaybi, B.; Moore-Olufemi, S.D.; Weisbrodt, N.W.; Moore, F.A. Peroxisome proliferator-activated receptor gamma mediates protection against cyclooxygenase-2-induced gut dysfunction in a rodent model of mesenteric ischemia/reperfusion. Shock 2005, 24, 462–469. [Google Scholar]
- Saubermann, L.J.; Nakajima, A.; Wada, K.; Zhao, S.; Terauchi, Y.; Kadowaki, T.; Aburatani, H.; Matsuhashi, N.; Nagai, R.; Blumberg, R.S. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm. Bowel Dis. 2002, 8, 330–339. [Google Scholar]
- Su, C.G.; Wen, X.; Bailey, S.T.; Jiang, W.; Rangwala, S.M.; Keilbaugh, S.A.; Flanigan, A.; Murthy, S.; Lazar, M.A.; Wu, G.D. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J. Clin. Investig. 1999, 104, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Kundu, P.; Ling, T.W.; Korecka, A.; Li, Y.; D’Arienzo, R.; Bunte, R.M.; Berger, T.; Arulampalam, V.; Chambon, P.; Mak, T.W.; et al. Absence of intestinal PPARgamma aggravates acute infectious colitis in mice through a lipocalin-2-dependent pathway. PLoS Pathog 2014, 10, e1003887. [Google Scholar] [CrossRef] [Green Version]
- Michalik, L.; Wahli, W. PPARs Mediate Lipid Signaling in Inflammation and Cancer. PPAR Res. 2008, 2008, 134059. [Google Scholar] [CrossRef] [Green Version]
- Shah, Y.M.; Morimura, K.; Gonzalez, F.J. Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G657–G666. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.; Hong, J.; Fraser, A.; Merriman, T.; Krissansen, G. PPAR-gamma and Crohn’s disease in New Zealand. Gastroenterology 2006, 130, 2249–2250. [Google Scholar] [CrossRef]
- Wada, K.; Nakajima, A.; Blumberg, R.S. PPARgamma and inflammatory bowel disease: A new therapeutic target for ulcerative colitis and Crohn’s disease. Trends Mol. Med. 2001, 7, 329–331. [Google Scholar]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [CrossRef]
- Gosset, P.; Charbonnier, A.S.; Delerive, P.; Fontaine, J.; Staels, B.; Pestel, J.; Tonnel, A.B.; Trottein, F. Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur. J. Immunol. 2001, 31, 2857–2865. [Google Scholar]
- Faveeuw, C.; Fougeray, S.; Angeli, V.; Fontaine, J.; Chinetti, G.; Gosset, P.; Delerive, P.; Maliszewski, C.; Capron, M.; Staels, B.; et al. Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 2000, 486, 261–266. [Google Scholar]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.; Schmidt, S.; Heun, R.; Klockgether, T.; Kolsch, H. Association of the PPARgamma gene polymorphism Pro12Ala with delayed onset of multiple sclerosis. Neurosci. Lett. 2009, 449, 81–83. [Google Scholar] [CrossRef]
- Regieli, J.J.; Jukema, J.W.; Doevendans, P.A.; Zwinderman, A.H.; van der Graaf, Y.; Kastelein, J.J.; Grobbee, D.E. PPAR gamma variant influences angiographic outcome and 10-year cardiovascular risk in male symptomatic coronary artery disease patients. Diabetes Care 2009, 32, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Xie, Z.; Guo, J.; Wang, Y.; Liu, C.; Zhang, S.; Tang, W.; Chen, Y. Association of PPARG rs 1801282 C>G polymorphism with risk of colorectal cancer: From a case-control study to a meta-analysis. Oncotarget 2017, 8, 100558–100569. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Chen, Y.; Tang, W.F.; Liu, C.; Zhang, S.; Guo, Z.Q.; Chen, G.; Zheng, X.W. PPARG rs3856806 C>T Polymorphism Increased the Risk of Colorectal Cancer: A Case-Control Study in Eastern Chinese Han Population. Front. Oncol. 2019, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Bassaganya-Riera, J.; Reynolds, K.; Martino-Catt, S.; Cui, Y.; Hennighausen, L.; Gonzalez, F.; Rohrer, J.; Benninghoff, A.U.; Hontecillas, R. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 2004, 127, 777–791. [Google Scholar]
- Yasui, Y.; Hosokawa, M.; Sahara, T.; Suzuki, R.; Ohgiya, S.; Kohno, H.; Tanaka, T.; Miyashita, K. Bitter gourd seed fatty acid rich in 9c,11t,13t-conjugated linolenic acid induces apoptosis and up-regulates the GADD45, p53 and PPARgamma in human colon cancer Caco-2 cells. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 113–119. [Google Scholar] [CrossRef]
- Akinyeke, T.O.; Stewart, L.V. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARgamma-independent mechanism. Cancer Biol. Ther. 2011, 11, 1046–1058. [Google Scholar]
- Yang, W.L.; Frucht, H. Activation of the PPAR pathway induces apoptosis and COX-2 inhibition in HT-29 human colon cancer cells. Carcinogenesis 2001, 22, 1379–1383. [Google Scholar]
- Subbaramaiah, K.; Lin, D.T.; Hart, J.C.; Dannenberg, A.J. Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J. Biol. Chem. 2001, 276, 12440–12448. [Google Scholar] [CrossRef] [Green Version]
- Vandoros, G.P.; Konstantinopoulos, P.A.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papachristou, G.I.; Karamouzis, M.V.; Gkermpesi, M.; Varakis, I.; Papavassiliou, A.G. PPAR-gamma is expressed and NF-kB pathway is activated and correlates positively with COX-2 expression in stromal myofibroblasts surrounding colon adenocarcinomas. J. Cancer Res. Clin. Oncol. 2006, 132, 76–84. [Google Scholar] [CrossRef]
- Heneka, M.T.; Klockgether, T.; Feinstein, D.L. Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J. Neurosci. 2000, 20, 6862–6867. [Google Scholar]
- Girnun, G.D.; Smith, W.M.; Drori, S.; Sarraf, P.; Mueller, E.; Eng, C.; Nambiar, P.; Rosenberg, D.W.; Bronson, R.T.; Edelmann, W.; et al. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc. Natl. Acad. Sci. USA 2002, 99, 13771–13776. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.; Nakajima, A.; Fujisawa, N.; Takahashi, H.; Ikeda, I.; Tomimoto, A.; Yonemitsu, K.; Nakajima, N.; Kudo, C.; Wada, K.; et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses colonic epithelial cell turnover and colon carcinogenesis through inhibition of the beta-catenin/T cell factor (TCF) pathway. J. Pharmacol. Sci. 2008, 106, 627–638. [Google Scholar]
- Peeters, A.; Baes, M. Role of PPAR in Hepatic Carbohydrate Metabolism. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Xiao, G.; Trujillo, C.; Chang, V.; Blanco, L.; Joseph, S.B.; Bassilian, S.; Saad, M.F.; Tontonoz, P.; Lee, W.N.; et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J. Biol. Chem. 2002, 277, 50237–50244. [Google Scholar] [CrossRef] [Green Version]
- Mandard, S.; Stienstra, R.; Escher, P.; Tan, N.S.; Kim, I.; Gonzalez, F.J.; Wahli, W.; Desvergne, B.; Muller, M.; Kersten, S. Glycogen synthase 2 is a novel target gene of peroxisome proliferator-activated receptors. Cell Mol. Life Sci. 2007, 64, 1145–1157. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Philp, A.; MacKenzie, M.G.; Belew, M.Y.; Towler, M.C.; Corstorphine, A.; Papalamprou, A.; Hardie, D.G.; Baar, K. Glycogen content regulates peroxisome proliferator activated receptor- partial differential (PPAR- partial differential) activity in rat skeletal muscle. PLoS ONE 2013, 8, e77200. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef] [Green Version]
- Louet, J.F.; Chatelain, F.; Decaux, J.F.; Park, E.A.; Kohl, C.; Pineau, T.; Girard, J.; Pegorier, J.P. Long-chain fatty acids regulate liver carnitine palmitoyltransferase I gene (L-CPT I) expression through a peroxisome-proliferator-activated receptor alpha (PPARalpha)-independent pathway. Biochem. J. 2001, 354, 189–197. [Google Scholar] [CrossRef]
- Mascaro, C.; Acosta, E.; Ortiz, J.A.; Marrero, P.F.; Hegardt, F.G.; Haro, D. Control of human muscle-type carnitine palmitoyltransferase I gene transcription by peroxisome proliferator-activated receptor. J. Biol. Chem. 1998, 273, 8560–8563. [Google Scholar] [CrossRef] [Green Version]
- Barrero, M.J.; Camarero, N.; Marrero, P.F.; Haro, D. Control of human carnitine palmitoyltransferase II gene transcription by peroxisome proliferator-activated receptor through a partially conserved peroxisome proliferator-responsive element. Biochem. J. 2003, 369, 721–729. [Google Scholar] [CrossRef]
- Gulick, T.; Cresci, S.; Caira, T.; Moore, D.D.; Kelly, D.P. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc. Natl. Acad. Sci. USA 1994, 91, 11012–11016. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Galman, C.; Lundasen, T.; Kharitonenkov, A.; Bina, H.A.; Eriksson, M.; Hafstrom, I.; Dahlin, M.; Amark, P.; Angelin, B.; Rudling, M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 2008, 8, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Regnier, M.; Fouche, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARalpha Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Brocker, C.N.; Yan, T.; Xie, C.; Krausz, K.W.; Xiang, R.; Gonzalez, F.J. Corrigendum to “Metabolic adaptation to intermittent fasting is independent of peroxisome proliferator-activated receptor alpha” [Mol Metab 7 (2018) 80–89]. Mol. Metab. 2018, 9, 217–219. [Google Scholar] [CrossRef]
- Chaix, A.; Lin, T.; Le, H.D.; Chang, M.W.; Panda, S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metab. 2019, 29, 303–319. [Google Scholar] [CrossRef]
- Adamo, K.B.; Dent, R.; Langefeld, C.D.; Cox, M.; Williams, K.; Carrick, K.M.; Stuart, J.S.; Sundseth, S.S.; Harper, M.E.; McPherson, R.; et al. Peroxisome proliferator-activated receptor gamma 2 and acyl-CoA synthetase 5 polymorphisms influence diet response. Obesity (Silver Spring) 2007, 15, 1068–1075. [Google Scholar] [CrossRef]
- Matsuo, T.; Nakata, Y.; Katayama, Y.; Iemitsu, M.; Maeda, S.; Okura, T.; Kim, M.K.; Ohkubo, H.; Hotta, K.; Tanaka, K. PPARG genotype accounts for part of individual variation in body weight reduction in response to calorie restriction. Obesity (Silver Spring) 2009, 17, 1924–1931. [Google Scholar] [CrossRef]
- Stryjecki, C.; Peralta-Romero, J.; Alyass, A.; Karam-Araujo, R.; Suarez, F.; Gomez-Zamudio, J.; Burguete-Garcia, A.; Cruz, M.; Meyre, D. Association between PPAR-gamma2 Pro12Ala genotype and insulin resistance is modified by circulating lipids in Mexican children. Sci. Rep. 2016, 6, 24472. [Google Scholar] [CrossRef] [Green Version]
- Masugi, J.; Tamori, Y.; Mori, H.; Koike, T.; Kasuga, M. Inhibitory effect of a proline-to-alanine substitution at codon 12 of peroxisome proliferator-activated receptor-gamma 2 on thiazolidinedione-induced adipogenesis. Biochem. Biophys. Res. Commun. 2000, 268, 178–182. [Google Scholar] [CrossRef]
- Memisoglu, A.; Hu, F.B.; Hankinson, S.E.; Manson, J.E.; De Vivo, I.; Willett, W.C.; Hunter, D.J. Interaction between a peroxisome proliferator-activated receptor gamma gene polymorphism and dietary fat intake in relation to body mass. Hum. Mol. Genet. 2003, 12, 2923–2929. [Google Scholar] [CrossRef] [Green Version]
- Robitaille, J.; Despres, J.P.; Perusse, L.; Vohl, M.C. The PPAR-gamma P12A polymorphism modulates the relationship between dietary fat intake and components of the metabolic syndrome: Results from the Quebec Family Study. Clin. Genet. 2003, 63, 109–116. [Google Scholar] [CrossRef]
- Luan, J.; Browne, P.O.; Harding, A.H.; Halsall, D.J.; O’Rahilly, S.; Chatterjee, V.K.; Wareham, N.J. Evidence for gene-nutrient interaction at the PPARgamma locus. Diabetes 2001, 50, 686–689. [Google Scholar] [CrossRef] [Green Version]
- Lindi, V.; Schwab, U.; Louheranta, A.; Laakso, M.; Vessby, B.; Hermansen, K.; Storlien, L.; Riccardi, G.; Matti IJ Uusitupa for the KANWU Study Group. Impact of the Pro12Ala polymorphism of the PPAR-gamma2 gene on serum triacylglycerol response to n-3 fatty acid supplementation. Mol. Genet. Metab. 2003, 79, 52–60. [Google Scholar] [CrossRef]
- Nicklas, B.J.; van Rossum, E.F.; Berman, D.M.; Ryan, A.S.; Dennis, K.E.; Shuldiner, A.R. Genetic variation in the peroxisome proliferator-activated receptor-gamma2 gene (Pro12Ala) affects metabolic responses to weight loss and subsequent weight regain. Diabetes 2001, 50, 2172–2176. [Google Scholar] [CrossRef] [Green Version]
- Ballor, D.L.; Poehlman, E.T. Exercise intensity does not affect depression of resting metabolic rate during severe diet restriction in male Sprague-Dawley rats. J. Nutr. 1993, 123, 1270–1276. [Google Scholar]
- Koizumi, A.; Wada, Y.; Tuskada, M.; Kayo, T.; Naruse, M.; Horiuchi, K.; Mogi, T.; Yoshioka, M.; Sasaki, M.; Miyamaura, Y.; et al. A tumor preventive effect of dietary restriction is antagonized by a high housing temperature through deprivation of torpor. Mech. Ageing Dev. 1996, 92, 67–82. [Google Scholar]
- Tabarean, I.; Morrison, B.; Marcondes, M.C.; Bartfai, T.; Conti, B. Hypothalamic and dietary control of temperature-mediated longevity. Ageing Res. Rev. 2010, 9, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Rachid, T.L.; Penna-de-Carvalho, A.; Bringhenti, I.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Fenofibrate (PPARalpha agonist) induces beige cell formation in subcutaneous white adipose tissue from diet-induced male obese mice. Mol. Cell Endocrinol. 2015, 402, 86–94. [Google Scholar] [CrossRef]
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-alpha and PPAR-beta/delta on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS ONE 2018, 13, e0191365. [Google Scholar] [CrossRef]
- Lu, C.; Cheng, S.Y. Thyroid hormone receptors regulate adipogenesis and carcinogenesis via crosstalk signaling with peroxisome proliferator-activated receptors. J. Mol. Endocrinol. 2010, 44, 143–154. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Brent, G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010, 21, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Colbert, L.H.; Mai, V.; Tooze, J.A.; Perkins, S.N.; Berrigan, D.; Hursting, S.D. Negative energy balance induced by voluntary wheel running inhibits polyp development in APCMin mice. Carcinogenesis 2006, 27, 2103–2107. [Google Scholar] [CrossRef] [Green Version]
- Holloszy, J.O. Mortality rate and longevity of food-restricted exercising male rats: A reevaluation. J. Appl. Physiol. (1985) 1997, 82, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Manini, T.M.; Everhart, J.E.; Patel, K.V.; Schoeller, D.A.; Colbert, L.H.; Visser, M.; Tylavsky, F.; Bauer, D.C.; Goodpaster, B.H.; Harris, T.B. Daily activity energy expenditure and mortality among older adults. JAMA 2006, 296, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Warburton, D.E.; Nicol, C.W.; Bredin, S.S. Health benefits of physical activity: The evidence. CMAJ 2006, 174, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Laaksonen, D.E.; Lindstrom, J.; Lakka, T.A.; Eriksson, J.G.; Niskanen, L.; Wikstrom, K.; Aunola, S.; Keinanen-Kiukaanniemi, S.; Laakso, M.; Valle, T.T.; et al. Physical activity in the prevention of type 2 diabetes: The Finnish diabetes prevention study. Diabetes 2005, 54, 158–165. [Google Scholar] [CrossRef]
- Holloszy, J.O. Exercise and longevity: Studies on rats. J. Gerontol. 1988, 43, B149–B151. [Google Scholar] [CrossRef]
- Samorajski, T.; Delaney, C.; Durham, L.; Ordy, J.M.; Johnson, J.A.; Dunlap, W.P. Effect of exercise on longevity, body weight, locomotor performance, and passive-avoidance memory of C57BL/6J mice. Neurobiol. Aging 1985, 6, 17–24. [Google Scholar] [CrossRef]
- Pekkanen, J.; Marti, B.; Nissinen, A.; Tuomilehto, J.; Punsar, S.; Karvonen, M.J. Reduction of premature mortality by high physical activity: A 20-year follow-up of middle-aged Finnish men. Lancet 1987, 1, 1473–1477. [Google Scholar] [CrossRef]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-term low-calorie low-protein vegan diet and endurance exercise are associated with low cardiometabolic risk. Rejuvenation Res. 2007, 10, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Klein, S.; Holloszy, J.O. Long-term low-protein, low-calorie diet and endurance exercise modulate metabolic factors associated with cancer risk. Am. J. Clin. Nutr. 2006, 84, 1456–1462. [Google Scholar] [CrossRef]
- Huffman, D.M.; Moellering, D.R.; Grizzle, W.E.; Stockard, C.R.; Johnson, M.S.; Nagy, T.R. Effect of exercise and calorie restriction on biomarkers of aging in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1618–R1627. [Google Scholar] [CrossRef]
- Thomas, A.W.; Davies, N.A.; Moir, H.; Watkeys, L.; Ruffino, J.S.; Isa, S.A.; Butcher, L.R.; Hughes, M.G.; Morris, K.; Webb, R. Exercise-associated generation of PPARgamma ligands activates PPARgamma signaling events and upregulates genes related to lipid metabolism. J. Appl. Physiol. 2012, 112, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, Y.; Li, Q.; Dong, X.; Hu, H.; Hu, R.; Ye, H.; Wu, Y.; Hu, R.; Li, Y. Exercise improved rat metabolism by raising PPAR-alpha. Int. J. Sports Med. 2011, 32, 568–573. [Google Scholar] [CrossRef]
- Santos, M.H.; Higuchi Mde, L.; Tucci, P.J.; Garavelo, S.M.; Reis, M.M.; Antonio, E.L.; Serra, A.J.; Maranhao, R.C. Previous exercise training increases levels of PPAR-alpha in long-term post-myocardial infarction in rats, which is correlated with better inflammatory response. Clinics (Sao Paulo) 2016, 71, 163–168. [Google Scholar] [CrossRef]
- Chen, W.; Gao, R.; Xie, X.; Zheng, Z.; Li, H.; Li, S.; Dong, F.; Wang, L. A metabolomic study of the PPARdelta agonist GW501516 for enhancing running endurance in Kunming mice. Sci. Rep. 2015, 5, 9884. [Google Scholar] [CrossRef] [Green Version]
- Butcher, L.R.; Thomas, A.; Backx, K.; Roberts, A.; Webb, R.; Morris, K. Low-intensity exercise exerts beneficial effects on plasma lipids via PPARgamma. Med. Sci. Sports Exerc. 2008, 40, 1263–1270. [Google Scholar] [CrossRef]
- Ruschke, K.; Fishbein, L.; Dietrich, A.; Kloting, N.; Tonjes, A.; Oberbach, A.; Fasshauer, M.; Jenkner, J.; Schon, M.R.; Stumvoll, M.; et al. Gene expression of PPARgamma and PGC-1alpha in human omental and subcutaneous adipose tissues is related to insulin resistance markers and mediates beneficial effects of physical training. Eur. J. Endocrinol. 2010, 162, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Nakata, R.; Inoue, H.; Shimizu, M.; Inoue, J.; Sato, R. Role of AMPK and PPARgamma1 in exercise-induced lipoprotein lipase in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1085–E1092. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.P.; Kulaputana, O.; Ghiu, I.A.; Brandauer, J.; Wohn, C.R.; Phares, D.A.; Shuldiner, A.R.; Hagberg, J.M. Endurance training-induced changes in the insulin response to oral glucose are associated with the peroxisome proliferator-activated receptor-gamma2 Pro12Ala genotype in men but not in women. Metabolism 2005, 54, 97–102. [Google Scholar] [CrossRef]
- Kahara, T.; Takamura, T.; Hayakawa, T.; Nagai, Y.; Yamaguchi, H.; Katsuki, T.; Katsuki, K.; Katsuki, M.; Kobayashi, K. PPARgamma gene polymorphism is associated with exercise-mediated changes of insulin resistance in healthy men. Metabolism 2003, 52, 209–212. [Google Scholar] [CrossRef] [Green Version]
- Adamo, K.B.; Sigal, R.J.; Williams, K.; Kenny, G.; Prud’homme, D.; Tesson, F. Influence of Pro12Ala peroxisome proliferator-activated receptor gamma2 polymorphism on glucose response to exercise training in type 2 diabetes. Diabetologia 2005, 48, 1503–1509. [Google Scholar] [CrossRef] [Green Version]
- Ostergard, T.; Ek, J.; Hamid, Y.; Saltin, B.; Pedersen, O.B.; Hansen, T.; Schmitz, O. Influence of the PPAR-gamma2 Pro12Ala and ACE I/D polymorphisms on insulin sensitivity and training effects in healthy offspring of type 2 diabetic subjects. Horm. Metab. Res. 2005, 37, 99–105. [Google Scholar] [CrossRef]
- Pan, W.; Liu, C.; Zhang, J.; Gao, X.; Yu, S.; Tan, H.; Yu, J.; Qian, D.; Li, J.; Bian, S.; et al. Association Between Single Nucleotide Polymorphisms in PPARA and EPAS1 Genes and High-Altitude Appetite Loss in Chinese Young Men. Front. Physiol. 2019, 10, 59. [Google Scholar] [CrossRef]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodriguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Fu, J.; Dipatrizio, N.V.; Guijarro, A.; Schwartz, G.J.; Li, X.; Gaetani, S.; Astarita, G.; Piomelli, D. Sympathetic activity controls fat-induced oleoylethanolamide signaling in small intestine. J. Neurosci. 2011, 31, 5730–5736. [Google Scholar] [CrossRef]
- Magotti, P.; Bauer, I.; Igarashi, M.; Babagoli, M.; Marotta, R.; Piomelli, D.; Garau, G. Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: Regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 2015, 23, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, S.; Oveisi, F.; Piomelli, D. Modulation of meal pattern in the rat by the anorexic lipid mediator oleoylethanolamide. Neuropsychopharmacology 2003, 28, 1311–1316. [Google Scholar] [CrossRef]
- Fu, J.; Oveisi, F.; Gaetani, S.; Lin, E.; Piomelli, D. Oleoylethanolamide, an endogenous PPAR-alpha agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 2005, 48, 1147–1153. [Google Scholar] [CrossRef]
- Caillon, A.; Duszka, K.; Wahli, W.; Rohner-Jeanrenaud, F.; Altirriba, J. The OEA effect on food intake is independent from the presence of PPARalpha in the intestine and the nodose ganglion, while the impact of OEA on energy expenditure requires the presence of PPARalpha in mice. Metabolism, 2018. [Google Scholar] [CrossRef]
- Lo Verme, J.; Gaetani, S.; Fu, J.; Oveisi, F.; Burton, K.; Piomelli, D. Regulation of food intake by oleoylethanolamide. Cell Mol. Life Sci. 2005, 62, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Piomelli, D. A fatty gut feeling. Trends Endocrinol. Metab. 2013, 24, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Proulx, K.; Cota, D.; Castaneda, T.R.; Tschop, M.H.; D’Alessio, D.A.; Tso, P.; Woods, S.C.; Seeley, R.J. Mechanisms of oleoylethanolamide-induced changes in feeding behavior and motor activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R729–R737. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez de Fonseca, F.; Navarro, M.; Gomez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodriguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An anorexic lipid mediator regulated by feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, S.; Fu, J.; Cassano, T.; Dipasquale, P.; Romano, A.; Righetti, L.; Cianci, S.; Laconca, L.; Giannini, E.; Scaccianoce, S.; et al. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. J. Neurosci. 2010, 30, 8096–8101. [Google Scholar] [CrossRef] [Green Version]
- Koleva, D.I.; Orbetzova, M.M.; Atanassova, P.K. Adipose tissue hormones and appetite and body weight regulators in insulin resistance. Folia Med. (Plovdiv) 2013, 55, 25–32. [Google Scholar]
- Gautron, L.; Laye, S. Neurobiology of inflammation-associated anorexia. Front. Neurosci. 2009, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Wang, X.; Li, Q.; Su, M.; Chew, E.; Wong, E.T.; Lacza, Z.; Radda, G.K.; Tergaonkar, V.; Han, W. Nuclear factor kappaB (NF-kappaB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia 2013, 56, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Jang, P.G.; Namkoong, C.; Kang, G.M.; Hur, M.W.; Kim, S.W.; Kim, G.H.; Kang, Y.; Jeon, M.J.; Kim, E.H.; Lee, M.S.; et al. NF-kappaB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. J. Biol. Chem. 2010, 285, 9706–9715. [Google Scholar] [CrossRef] [Green Version]
- Speakman, J.R. Body size, energy metabolism and lifespan. J. Exp. Biol. 2005, 208, 1717–1730. [Google Scholar] [CrossRef] [Green Version]
- Pearl, R. The Rate of Living; University of London Press: London, UK, 1928. [Google Scholar]
- Houthoofd, K.; Braeckman, B.P.; Lenaerts, I.; Brys, K.; De Vreese, A.; Van Eygen, S.; Vanfleteren, J.R. Axenic growth up-regulates mass-specific metabolic rate, stress resistance, and extends life span in Caenorhabditis elegans. Exp. Gerontol. 2002, 37, 1371–1378. [Google Scholar] [CrossRef]
- Metaxakis, A.; Partridge, L. Dietary restriction extends lifespan in wild-derived populations of Drosophila melanogaster. PLoS ONE 2013, 8, e74681. [Google Scholar] [CrossRef]
- Abalan, F.; Mayo, W.; Simon, H.; Le Moal, M. Paradoxical effect of severe dietary restriction on Long-Evans rat life span. Int. J. Vitam. Nutr. Res. 2010, 80, 386–393. [Google Scholar] [CrossRef]
- Pifferi, F.; Terrien, J.; Marchal, J.; Dal-Pan, A.; Djelti, F.; Hardy, I.; Chahory, S.; Cordonnier, N.; Desquilbet, L.; Hurion, M.; et al. Caloric restriction increases lifespan but affects brain integrity in grey mouse lemur primates. Commun. Biol. 2018, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef]
- Knight, J.A. The biochemistry of aging. Adv. Clin. Chem. 2000, 35, 1–62. [Google Scholar]
- Park, S.Y.; Kim, Y.W.; Kim, J.E.; Kim, J.Y. Age-associated changes in fat metabolism in the rat and its relation to sympathetic activity. Life Sci. 2006, 79, 2228–2233. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism-novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Cock, T.A.; Houten, S.M.; Auwerx, J. Peroxisome proliferator-activated receptor-gamma: Too much of a good thing causes harm. EMBO Rep. 2004, 5, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Argmann, C.; Dobrin, R.; Heikkinen, S.; Auburtin, A.; Pouilly, L.; Cock, T.A.; Koutnikova, H.; Zhu, J.; Schadt, E.E.; Auwerx, J. Ppargamma2 is a key driver of longevity in the mouse. PLoS Genet. 2009, 5, e1000752. [Google Scholar] [CrossRef]
- Viguerie, N.; Vidal, H.; Arner, P.; Holst, C.; Verdich, C.; Avizou, S.; Astrup, A.; Saris, W.H.; Macdonald, I.A.; Klimcakova, E.; et al. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia 2005, 48, 123–131. [Google Scholar] [CrossRef]
- Karbowska, J.; Kochan, Z. Intermittent fasting up-regulates Fsp27/Cidec gene expression in white adipose tissue. Nutrition 2012, 28, 294–299. [Google Scholar] [CrossRef]
- Lee, C.K.; Allison, D.B.; Brand, J.; Weindruch, R.; Prolla, T.A. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc. Natl. Acad. Sci. USA 2002, 99, 14988–14993. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.; Youssef, J.; Rezaiekhaleigh, M.; Birnbaum, L.S.; Badr, M. Senescence-associated decline in hepatic peroxisomal enzyme activities corresponds with diminished levels of retinoid X receptor alpha, but not peroxisome proliferator-activated receptor alpha. Mech. Ageing Dev. 2002, 123, 1469–1476. [Google Scholar] [CrossRef]
- Long, X.; Boluyt, M.O.; O’Neill, L.; Zheng, J.S.; Wu, G.; Nitta, Y.K.; Crow, M.T.; Lakatta, E.G. Myocardial retinoid X receptor, thyroid hormone receptor, and myosin heavy chain gene expression in the rat during adult aging. J. Gerontol. A Biol. Sci. Med. Sci. 1999, 54, B23–B27. [Google Scholar] [CrossRef] [Green Version]
- Enderlin, V.; Pallet, V.; Alfos, S.; Dargelos, E.; Jaffard, R.; Garcin, H.; Higueret, P. Age-related decreases in mRNA for brain nuclear receptors and target genes are reversed by retinoic acid treatment. Neurosci. Lett. 1997, 229, 125–129. [Google Scholar] [CrossRef]
- Stauber, A.J.; Brown-Borg, H.; Liu, J.; Waalkes, M.P.; Laughter, A.; Staben, R.A.; Coley, J.C.; Swanson, C.; Voss, K.A.; Kopchick, J.J.; et al. Constitutive expression of peroxisome proliferator-activated receptor alpha-regulated genes in dwarf mice. Mol. Pharmacol. 2005, 67, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xie, Y.; Berglund, E.D.; Coate, K.C.; He, T.T.; Katafuchi, T.; Xiao, G.; Potthoff, M.J.; Wei, W.; Wan, Y.; et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. Elife 2012, 1, e00065. [Google Scholar] [CrossRef]
- Olsson, B.; Bohlooly, Y.M.; Brusehed, O.; Isaksson, O.G.; Ahren, B.; Olofsson, S.O.; Oscarsson, J.; Tornell, J. Bovine growth hormone-transgenic mice have major alterations in hepatic expression of metabolic genes. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E504–E511. [Google Scholar] [CrossRef]
- Sugiyama, H.; Yamada, J.; Suga, T. Effects of testosterone, hypophysectomy and growth hormone treatment on clofibrate induction of peroxisomal beta-oxidation in female rat liver. Biochem. Pharmacol. 1994, 47, 918–921. [Google Scholar] [CrossRef]
- Zhou, Y.C.; Waxman, D.J. Cross-talk between janus kinase-signal transducer and activator of transcription (JAK-STAT) and peroxisome proliferator-activated receptor-alpha (PPARalpha) signaling pathways. Growth hormone inhibition of pparalpha transcriptional activity mediated by stat5b. J. Biol. Chem. 1999, 274, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.C.; Waxman, D.J. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain. J. Biol. Chem. 1999, 274, 29874–29882. [Google Scholar] [CrossRef] [Green Version]
- Shipley, J.M.; Waxman, D.J. Down-regulation of STAT5b transcriptional activity by ligand-activated peroxisome proliferator-activated receptor (PPAR) alpha and PPARgamma. Mol. Pharmacol. 2003, 64, 355–364. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, S.; Jia, W. Calorie restriction and its impact on gut microbial composition and global metabolism. Front. Med. 2018, 12, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, A.; Cerdo, T.; Jauregui, R.; Pieper, D.H.; Marcos, A.; Clemente, A.; Garcia, F.; Margolles, A.; Ferrer, M.; Campoy, C.; et al. One-year calorie restriction impacts gut microbial composition but not its metabolic performance in obese adolescents. Environ. Microbiol. 2017, 19, 1536–1551. [Google Scholar] [CrossRef]
- Tanca, A.; Abbondio, M.; Palomba, A.; Fraumene, C.; Marongiu, F.; Serra, M.; Pagnozzi, D.; Laconi, E.; Uzzau, S. Caloric restriction promotes functional changes involving short-chain fatty acid biosynthesis in the rat gut microbiota. Sci. Rep. 2018, 8, 14778. [Google Scholar] [CrossRef]
- Zhang, C.; Li, S.; Yang, L.; Huang, P.; Li, W.; Wang, S.; Zhao, G.; Zhang, M.; Pang, X.; Yan, Z.; et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 2013, 4, 2163. [Google Scholar] [CrossRef] [Green Version]
- Fraumene, C.; Manghina, V.; Cadoni, E.; Marongiu, F.; Abbondio, M.; Serra, M.; Palomba, A.; Tanca, A.; Laconi, E.; Uzzau, S. Caloric restriction promotes rapid expansion and long-lasting increase of Lactobacillus in the rat fecal microbiota. Gut Microbes 2018, 9, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wu, Y.; Wang, L.; Pang, X.; Zhao, L.; Yuan, H.; Zhang, C. A More Robust Gut Microbiota in Calorie-Restricted Mice Is Associated with Attenuated Intestinal Injury Caused by the Chemotherapy Drug Cyclophosphamide. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Huang, M.; You, X.; Zhao, J.; Chen, L.; Wang, L.; Luo, Y.; Chen, Y. Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 2018, 8, 13037. [Google Scholar] [CrossRef] [Green Version]
- Crawford, P.A.; Crowley, J.R.; Sambandam, N.; Muegge, B.D.; Costello, E.K.; Hamady, M.; Knight, R.; Gordon, J.I. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 11276–11281. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Korecka, A.; Polizzi, A.; Lippi, Y.; Blum, Y.; Canlet, C.; Tremblay-Franco, M.; Gautier-Stein, A.; Burcelin, R.; Yen, Y.C.; et al. Hepatic circadian clock oscillators and nuclear receptors integrate microbiome-derived signals. Sci. Rep. 2016, 6, 20127. [Google Scholar] [CrossRef]
- Mukherji, A.; Kobiita, A.; Ye, T.; Chambon, P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 2013, 153, 812–827. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARalpha Signaling Limits Inflammatory Responses to Commensal Microbiota in the Intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef]
- Li, G.; Xie, C.; Lu, S.; Nichols, R.G.; Tian, Y.; Li, L.; Patel, D.; Ma, Y.; Brocker, C.N.; Yan, T.; et al. Intermittent Fasting Promotes White Adipose Browning and Decreases Obesity by Shaping the Gut Microbiota. Cell Metab. 2017, 26, 801. [Google Scholar] [CrossRef] [Green Version]
- Burdick, A.D.; Kim, D.J.; Peraza, M.A.; Gonzalez, F.J.; Peters, J.M. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell. Signal. 2006, 18, 9–20. [Google Scholar] [CrossRef]
- Murakami, M.; Tognini, P.; Liu, Y.; Eckel-Mahan, K.L.; Baldi, P.; Sassone-Corsi, P. Gut microbiota directs PPARgamma-driven reprogramming of the liver circadian clock by nutritional challenge. EMBO Rep. 2016, 17, 1292–1303. [Google Scholar] [CrossRef] [Green Version]
- Peyrin-Biroulet, L.; Beisner, J.; Wang, G.; Nuding, S.; Oommen, S.T.; Kelly, D.; Parmentier-Decrucq, E.; Dessein, R.; Merour, E.; Chavatte, P.; et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc. Natl. Acad. Sci. USA 2010, 107, 8772–8777. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.Y.P.; Visvalingam, V.; Wahli, W. The PPAR-microbiota-metabolic organ trilogy to fine-tune physiology. FASEB J. 2019, 33, 9706–9730. [Google Scholar] [CrossRef] [Green Version]
- Fann, D.Y.; Ng, G.Y.; Poh, L.; Arumugam, T.V. Positive effects of intermittent fasting in ischemic stroke. Exp. Gerontol. 2017, 89, 93–102. [Google Scholar] [CrossRef]
- Razeghi Jahromi, S.; Ghaemi, A.; Alizadeh, A.; Sabetghadam, F.; Moradi Tabriz, H.; Togha, M. Effects of Intermittent Fasting on Experimental Autoimune Encephalomyelitis in C57BL/6 Mice. Iran. J. Allergy Asthma Immunol. 2016, 15, 212–219. [Google Scholar]
- Okada, T.; Otsubo, T.; Hagiwara, T.; Inazuka, F.; Kobayashi, E.; Fukuda, S.; Inoue, T.; Higuchi, K.; Kawamura, Y.I.; Dohi, T. Intermittent fasting prompted recovery from dextran sulfate sodium-induced colitis in mice. J. Clin. Biochem. Nutr. 2017, 61, 100–107. [Google Scholar] [CrossRef]
- Savendahl, L.; Underwood, L.E.; Haldeman, K.M.; Ulshen, M.H.; Lund, P.K. Fasting prevents experimental murine colitis produced by dextran sulfate sodium and decreases interleukin-1 beta and insulin-like growth factor I messenger ribonucleic acid. Endocrinology 1997, 138, 734–740. [Google Scholar] [CrossRef]
- Cheng, C.W.; Villani, V.; Buono, R.; Wei, M.; Kumar, S.; Yilmaz, O.H.; Cohen, P.; Sneddon, J.B.; Perin, L.; Longo, V.D. Fasting-Mimicking Diet Promotes Ngn3-Driven beta-Cell Regeneration to Reverse Diabetes. Cell 2017, 168, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Rangan, P.; Choi, I.; Wei, M.; Navarrete, G.; Guen, E.; Brandhorst, S.; Enyati, N.; Pasia, G.; Maesincee, D.; Ocon, V.; et al. Fasting-Mimicking Diet Modulates Microbiota and Promotes Intestinal Regeneration to Reduce Inflammatory Bowel Disease Pathology. Cell Rep. 2019, 26, 2704–2719. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Liu, B.; Heilbronn, L.K. Intermittent fasting: What questions should we be asking? Physiol. Behav. 2020, 218, 112827. [Google Scholar] [CrossRef]
- Horne, B.D.; Muhlestein, J.B.; Anderson, J.L. Health effects of intermittent fasting: Hormesis or harm? A systematic review. Am. J. Clin. Nutr. 2015, 102, 464–470. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. https://doi.org/10.3390/cells9071708
Duszka K, Gregor A, Guillou H, König J, Wahli W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity. Cells. 2020; 9(7):1708. https://doi.org/10.3390/cells9071708
Chicago/Turabian StyleDuszka, Kalina, András Gregor, Hervé Guillou, Jürgen König, and Walter Wahli. 2020. "Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity" Cells 9, no. 7: 1708. https://doi.org/10.3390/cells9071708
APA StyleDuszka, K., Gregor, A., Guillou, H., König, J., & Wahli, W. (2020). Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity. Cells, 9(7), 1708. https://doi.org/10.3390/cells9071708