Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development—A Focus on Ulcerative Colitis




Abstract
:1. Introduction
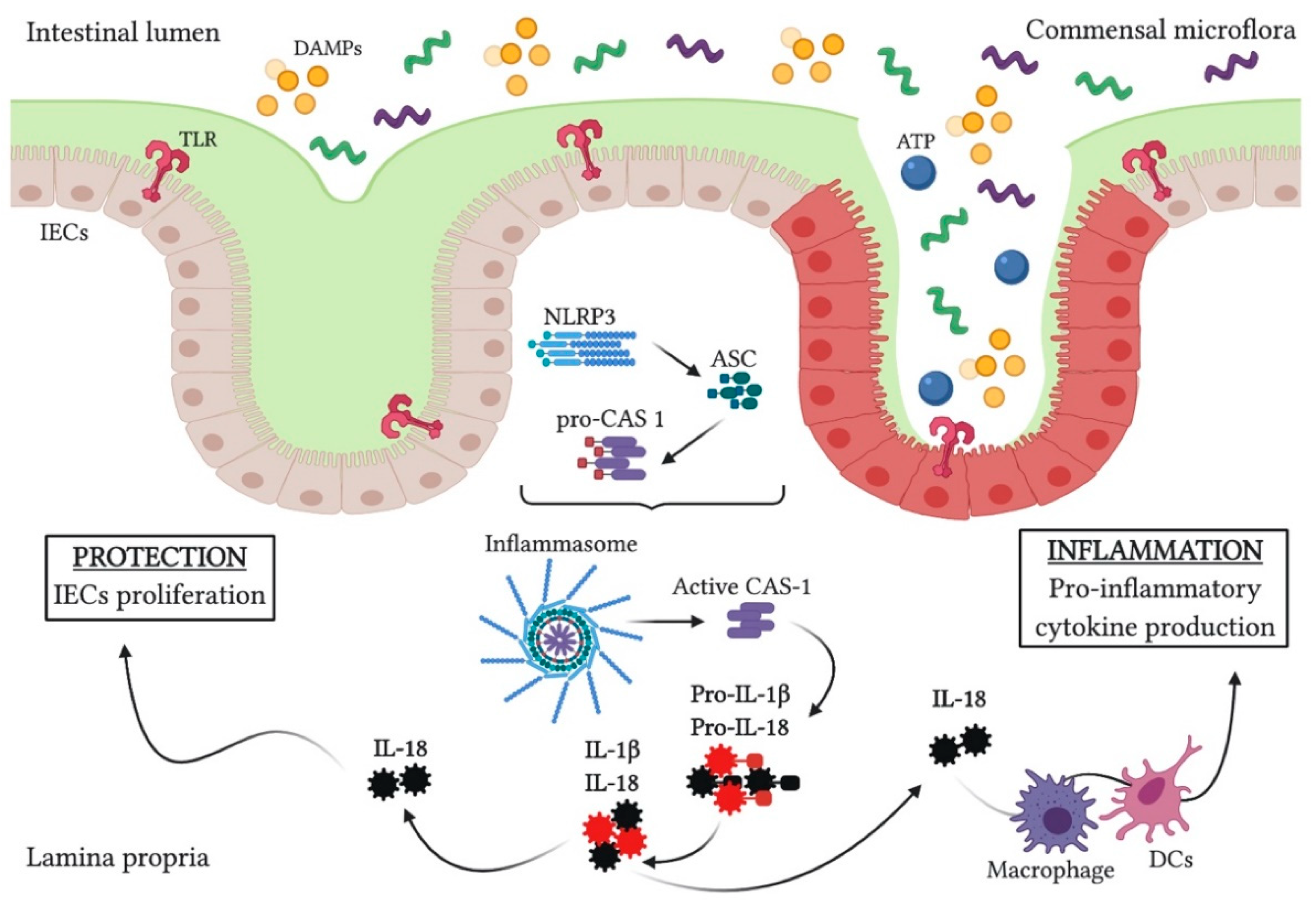
2. Inflammasomes
2.1. Molecular Mechanisms of NLRP3 Inflammasome Signaling
2.2. NLRP3 Inflammasome Activation in Ulcerative Colitis
3. Autophagy
3.1. Macroautophagy Controls NLRP3 Inflammasome Activation in Ulcerative Colitis Conditions
3.2. Controlling Mitochondrial Damage and Mitochondrial ROS throughout Mitophagy in UC
4. Endoplasmic Reticulum Stress and Unfolded Protein Response
4.1. Endoplasmic Reticulum Unfolded Protein Response
4.2. Mitochondrial Unfolded Protein Response
5. Transcriptome Profiling of Non-Coding Regions in Genetic Susceptibility Loci
6. Discussions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef]
- Lichtenstein, G.R.; Kamm, M.A. 5-aminosalicylate formulations for the treatment of ulcerative colitis—Methods of comparing release rates and delivery of 5-aminosalicylate to the colonic mucosa. Aliment. Pharmacol. Ther. 2008, 28, 663–673. [Google Scholar] [CrossRef]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef]
- Allocca, M.; Furfaro, F.; Fiorino, G.; Gilardi, D.; D’Alessio, S.; Danese, S. Can IL-23 be a good target for ulcerative colitis? Best Pract. Res. Clin. Gastroenterol. 2018, 32, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.H.; Zhu, C.X.; Quan, Y.S.; Yang, Z.Y.; Wu, S.; Luo, W.W.; Tan, B.; Wang, X.Y. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J. Gastroenterol. 2018, 24, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Tatiya-aphiradee, N.; Chatuphonprasert, W.; Jarukamjorn, K. Immune response and inflammatory pathway of ulcerative colitis. J. Basic Clin. Physiol. Pharmacol. 2018, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, H. NLRP3 inflammasome and inflammatory bowel disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Blumberg, R.S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011, 140, 1738–1747. [Google Scholar] [CrossRef] [Green Version]
- Randall-Demllo, S.; Chieppa, M.; Eri, R.D. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef] [Green Version]
- Coskun, M.; Bjerrum, J.T.; Seidelin, J.B.; Nielsen, O.H. MicroRNAs in inflammatory bowel disease-pathogenesis, diagnostics and therapeutics. World J. Gastroenterol. 2012, 18, 4629–4634. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Zhang, H.J. miRNAs as new molecular insights into inflammatory bowel disease: Crucial regulators in autoimmunity and inflammation. World J. Gastroenterol. 2016, 22, 2206–2218. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, A.; Hayward, J.A.; Man, S.M. Molecular mechanisms of inflammasome signaling. J. Leukoc. Biol. 2018, 103, 233–257. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Wang, S.L.; Pan, P.; Yao, J.; Wu, K.; Li, Z.S.; Bai, Y.; Linghu, E.Q. Targeting NLRP3 inflammasome in inflammatory bowel disease: Putting out the fire of inflammation. Inflammation 2019, 1–13. [Google Scholar] [CrossRef]
- Mehto, S.; Chauhan, S.; Jena, K.K.; Chauhan, N.R.; Nath, P.; Sahu, R.; Dhar, K.; Das, S.K.; Chauhan, S. IRGM restrains NLRP3 inflammasome activation by mediating its SQSTM1/p62-dependent selective autophagy. Autophagy 2019, 15, 1645–1647. [Google Scholar] [CrossRef]
- Lorenz, G.; Darisipudi, M.N.; Anders, H.J. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol. Dial. Transplant. 2014, 29, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourkochristou, E.; Aggeletopoulou, I.; Konstantakis, C.; Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J. Gastroenterol. 2019, 25, 4796–4804. [Google Scholar] [CrossRef]
- Loher, F.; Bauer, C.; Landauer, N.; Schmall, K.; Siegmund, B.; Lehr, H.A.; Dauer, M.; Schoenharting, S.E.; Eigler, A. The interleukin-1β-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J. Pharmacol. Exp. Ther. 2004, 308, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, M.; Darby, T.; Melgar, S. The complex role of inflammasomes in the pathogenesis of inflammatory bowel diseases–lessons learned from experimental models. Cytokine Growth Factor Rev. 2014, 25, 715–730. [Google Scholar] [CrossRef]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [Green Version]
- Lissner, D.; Siegmund, B. The multifaceted role of the inflammasome in inflammatory bowel diseases. Sci. World J. 2011, 11, 1536–1547. [Google Scholar] [CrossRef]
- Zambetti, L.P.; Mortellaro, A. NLRPs, microbiota, and gut homeostasis: Unravelling the connection. J. Pathol. 2014, 233, 321–330. [Google Scholar] [CrossRef]
- Stehlik, C.; Fiorentino, L.; Dorfleutner, A.; Bruey, J.M.; Ariza, E.M.; Sagara, J.; Reed, J.C. The PAAD/PYRIN-family protein ASC is a dual regulator of a conserved step in nuclear factor κB activation pathways. J. Exp. Med. 2002, 196, 1605–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Abraham, C. Inflammatory bowel disease genetics: Nod2. Annu. Rev. Med. 2007, 58, 401–416. [Google Scholar] [CrossRef]
- Freire, P.; Cardoso, R.; Figueiredo, P.; Donato, M.M.; Ferreira, M.; Mendes, S.; Ferreira, A.M.; Vasconcelos, H.; Portela, F.; Sofia, C. NOD2 gene mutations in ulcerative colitis: Useless or misunderstood? Int. J. Colorectal Dis. 2014, 29, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaki, M.H.; Lamkanfi, M.; Kanneganti, T.D. The Nlrp3 inflammasome in IBD and colorectal tumorigenesis. Trends Immunol. 2011, 32, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261–e267. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ye, Q.; Zeng, X.; Qiao, S. Functions of Macrophages in the Maintenance of Intestinal Homeostasis. J. Immunol. Res. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Haq, S.; Grondin, J.; Banskota, S.; Khan, W.I. Autophagy: Roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci. 2019, 26, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Pavel, M.; Rubinsztein, D.C. Mammalian autophagy and the plasma membrane. FEBS J. 2017, 284, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation: A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Palomino-Morales, R.J.; Oliver, J.; Gomez-Garcia, M.; Lopez-Nevot, M.A.; Rodrigo, L.; Nieto, A.; Alizadeh, B.Z.; Martin, J. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: A meta-analysis approach. Genes Immun. 2009, 10, 356–364. [Google Scholar] [CrossRef]
- Cheng, J.F.; Ning, Y.J.; Zhang, W.; Lu, Z.H.; Lin, L. T300A polymorphism of ATG16L1 and susceptibility to inflammatory bowel diseases: A meta-analysis. World J. Gastroenterol. 2010, 16, 1258–1266. [Google Scholar] [CrossRef]
- Nuij, V.J.A.A.; Peppelenbosch, M.P.; van der Woude, C.J.; Fuhler, G.M. Genetic polymorphism in ATG16L1 gene is associated with adalimumab use in inflammatory bowel disease. J. Transl. Med. 2017, 15, 248. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.; Lang, T.; Thomas, J.P.; Sukkar, M.B.; Nabar, N.R.; Kehrl, J.H. Autophagy and inflammasomes. Mol. Immunol. 2017, 86, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Münz, C. Regulation of innate immunity by the molecular machinery of macroautophagy. Cell. Microbiol. 2014, 16, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autop-hagy. Mol. Cell 2019, 73, 429–445. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Wang, Y.; Long, Z.; He, G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. 2019, 51, 1087–1095. [Google Scholar]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kabi, A.; McDonald, C. Role of Autophagy-Related Genes in the Pathology of Inflammatory Bowel Disease. eLS 2013. [Google Scholar] [CrossRef]
- Söderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzen, L.E.; Lindmark, T.; Wirén, M.; Tagesson, C.; Sjödahl, R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 2002, 50, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenburg, W.; Keijer, J.; Kramer, E.; Vink, C.; van der Meer, R.; Bovee-Oudenhoven, I.M. Impaired barrier function by dietary fructo-oligosaccharides (FOS) in rats is accompanied by increased colonic mitochondrial gene expression. BMC Genomics 2008, 9, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochnicki, T.; Latz, E. Inflammasomes on the cross-roads of innate immune recognition and metabolic control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Boyapati, R.K.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.; Deen, N.; Zamani, S.; Hasnat, M.A. Mitophagy and the release of inflammatory cytokines. Mitochondrion 2018, 41, 2–8. [Google Scholar] [CrossRef]
- Hauptmann, N.; Grimsby, J.; Shih, J.C.; Cadenas, E. The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch. Biochem. Biophys. 1996, 335, 295–304. [Google Scholar] [CrossRef]
- Corridoni, D.; Chapman, T.; Ambrose, T.; Simmons, A. Emerging mechanisms of innate immunity and their translational potential in inflammatory bowel disease. Front. Med. 2018, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Nishikawa, M.; Oshitani, N.; Matsumoto, T.; Nishigami, T.; Arakawa, T.; Inoue, M. Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br. J. Cancer 2005, 93, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozza, A.; Scarpa, M.; Ruffolo, C.; Polese, L.; Erroi, F.; Bridda, A.; Norberto, L.; Frego, M. Colonic carcinogenesis in IBD: Molecular events. Ann. Ital. Chir. 2011, 82, 19–28. [Google Scholar]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Kaser, A.; Martínez-Naves, E.; Blumberg, R.S. Endoplasmic reticulum stress: Implications for inflammatory bowel disease pathogenesis. Curr. Opin. Gastroenterol. 2010, 26, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Roberts, C.J. Non-native protein aggregation kinetics. Biotechnol. Bioeng. 2017, 98, 927–938. [Google Scholar] [CrossRef]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederreiter, L.; Kaser, A. Endoplasmic reticulum stress and inflammatory bowel disease. Acta Gastro-Ent. Belg. 2011, 74, 330–333. [Google Scholar]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef]
- Yaman, I.; Fernandez, J.; Liu, H.; Caprara, M.; Komar, A.A.; Koromalis, A.E.; Zhou, L.; Snider, M.D.; Scheuner, D.; Kaufman, R.J.; et al. The zipper model of translational control: A small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 2003, 113, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S. Epithelial ER stress in Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2016, 22, 984–993. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.S.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Adolph, T.E.; Blumberg, R.S. The unfolded protein response and gastrointestinal disease. Semin. Immunopathol. 2013, 35, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [Green Version]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Ron, D. The mitochondrial UPR–protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, C.J.; Wei, Z.; Li, J.; Zhu, J.; Gu, M.; Baldassano, R.N.; Grant, S.F.A.; Hakonarson, H. Transcriptome profiling of human ulcerative colitis mucosa reveals altered expression of pathways enriched in genetic susceptibility loci. PLoS ONE 2014, 9, e96153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharopoulou, E.; Gazouli, M.; Tzouvala, M.; Vezakis, A.; Karamanolis, G. The contribution of long non-coding RNAs in Inflammatory Bowel Diseases. Dig. Liver Dis. 2017, 49, 1067–1072. [Google Scholar] [CrossRef]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Li, L.; Cui, J.; Zhang, H.; Liu, Y.; Zhang, C.Y.; Zen, K. Role of miR-150-targeting c-Myb in colonic epithelial disruption during dextran sulphate sodium-induced murine experimental colitis and human ulcerative colitis. J. Pathol. 2011, 225, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, Y.; Shi, C.; Chen, H.; Zhang, H.; Chen, N.; Zhang, P.; Wang, F.; Yang, J.; Yang, J.; et al. Overexpression of miR-21 in patients with ulcerative colitis impairs intestinal epithelial barrier function through targeting the Rho GTPase RhoB. Biochem. Biophys. Res. Commun. 2013, 434, 746–752. [Google Scholar] [CrossRef]
- Feng, X.; Wang, H.; Ye, S.; Guan, J.; Tan, W.; Cheng, S.; Wei, G.; Wu, W.; Wu, F.; Zhou, Y. Up-regulation of microRNA-126 may contribute to pathogenesis of ulcerative colitis via regulating NF-kappaB inhibitor IκBα. PLoS ONE 2012, 7, e52782. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Banan, A.; Forsyth, C.B.; Fields, J.Z.; Lau, C.K.; Zhang, L.J.; Keshavarzian, A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2008, 32, 355–364. [Google Scholar] [CrossRef]
- Adammek, M.; Greve, B.; Kässens, N.; Schneider, C.; Brüggemann, K.; Schüring, A.N.; Starzinski-Powitz, A.; Kiesel, L.; Götte, M. MicroRNA miR-145 inhibits proliferation, invasiveness, and stem cell phenotype of an in vitro endometriosis model by targeting multiple cytoskeletal elements and pluripotency factors. Fertil. Steril. 2013, 99, 1346–1355. [Google Scholar] [CrossRef]
- Qiao, Y.Q.; Huang, M.L.; Xu, A.T.; Zhao, D.; Ran, Z.H.; Shen, J. LncRNA DQ786243 affects Treg related CREB and Foxp3 expression in Crohn’s disease. J. Biomed. Sci. 2013, 20, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.H.; Berthelsen, C.H.; Seemann, S.E.; Pan, X.; Frederiksen, K.S.; Vilien, M.; Gorodkin, J.; Pociot, F. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Med. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padua, D.; Mahurkar-Joshi, S.; Law, I.K.M.; Polytarchou, C.; Vu, J.P.; Pisegna, J.R.; Shih, D.; Iliopoulos, D.; Pothoulakis, C. A long noncoding RNA signature for ulcerative colitis identifies IFNG-AS1 as an enhancer of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G446–G457. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Huang, Y.; Dong, F.; Kwon, J.H. Ulcerative Colitis-Associated Long Noncoding RNA, BC012900, Regulates Intestinal Epithelial Cell Apoptosis. Inflamm. Bowel Dis. 2016, 22, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C.; Iliopoulos, D.; Rankin, R.; Padua, D. P-307 the long non-coding RNA, CDKN2B-AS1, Is associated with IBD and is downregulated by TGF-beta. Inflamm. Bowel Dis. 2017, 23. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Yorimitsu, T.; Klionsky, D.J. Eating the endoplasmic reticulum: Quality control by autophagy. Trends Cell Biol. 2007, 17, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Ron, D. Alterations in an IRE1-RNA complex in the mammalian unfolded protein response. J. Cell Sci. 2001, 114, 3207–3212. [Google Scholar] [PubMed]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- Majors, A.K.; Austin, R.C.; Carol, A.; Pyeritz, R.E.; Hascall, V.C.; Kessler, S.P.; Sen, G.; Strong, S.A. Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J. Biol. Chem. 2003, 278, 47223–47231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Nves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hrtwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabat, A.M.; Harrison, O.J.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.F.; Laing, A.; Abeler-Dorner, L.; Forman, S.P.; Grencis, R.K.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife 2016, 5, e12444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, L.; McGovern, D.P.B.; Hamill, A.M.; Ichikawa, R.; Kanazawa, Y.; Luu, J.; Kumegai, K.; Cilluffo, M.; Fukata, M.; et al. Myeloid ATG16L1 facilitates host–bacteria interactions in maintaining intestinal homeostasis. J. Immunol. 2017, 198, 2133–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szbadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase Kinase-β, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.I.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroku, Y.; Fujita, E.; Ueno, T.; Isoai, A.; Kumegai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2a phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Piao, J.H.; Nakajima, A.; Sakon-Komazawa, S.; Kojima, Y.; Mori, K.; Yagita, H.; Okumura, K.; Harding, H.; Nakano, H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 2005, 280, 33917–33925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Li, S.; Cai, L.; Liu, J.L.; Wang, C.Y.; Gan, W.J.; Li, X.M.; Wang, J.R.; Sun, L.N.; Deng, M.; et al. Erbin exerts a protective effect against inflammatory bowel disease by suppressing autophagic cell death. Oncotarget 2018, 9, 12035–12049. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Huang, J.; Brumell, J.H. NADPH oxidases contribute to autophagy regulation. Autophagy 2009, 5, 887–889. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, J. microRNAs as therapeutic targets in intestinal diseases. ExRNA 2019, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Valmiki, S.; Ahuja, V.; Paul, J. MicroRNA exhibit altered expression in the inflamed colonic mucosa of ulcerative colitis patients. World J. Gastroenterol. 2017, 23, 5324–5332. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Ramasamy, K.; Bouamar, H.; Lin, A.P.; Jiang, D.; Aguiar, R.C. MicroRNAs miR-125a and miR-125b constitutively activate the NF-κB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc. Natl. Acad. Sci. USA 2012, 109, 7865–7870. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Li, Y.; Shao, T.; Zhao, Z.; Wang, Y.; Wu, A.; Chen, H.; Li, S.; Jiang, C.; Xu, J.; et al. Integrating analysis reveals microRNA-mediated pathway crosstalk among Crohn’s disease, ulcerative colitis and colorectal cancer. Mol. Biosyst. 2014, 10, 2317–2328. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| microRNA | Expression | Type of Disease | Sample Type |
|---|---|---|---|
| miR-16 | Upregulated | Active UC vs. control | Sigmoid colon biopsies |
| miR-21 | Upregulated | Active UC vs. control | Sigmoid colon biopsies |
| miR-23a | Upregulated | Active/Inactive UC vs. control | Sigmoid colon biopsies |
| miR-24 | Upregulated | Active UC vs. control | Sigmoid colon biopsies |
| miR-29a | Upregulated | Active/Inactive UC vs. control | Sigmoid colon biopsies |
| miR-126 | Upregulated | Active UC vs. control | Sigmoid colon biopsies |
| miR-138 | Upregulated | Active UC vs. inactive UC | Colon biopsies |
| miR-150 | Upregulated | Active UC vs. inactive UC | Colon biopsies |
| miR-192 | Downregulated | Active UC vs. control | Peripheral blood |
| miR-212 | Downregulated | Inactive UC vs. control | Colon biopsies |
| miR-375 | Downregulated | Active UC vs. control | Colon biopsies |
| miR-422b | Upregulated | Active UC vs. control | Peripheral blood |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samoilă, I.; Dinescu, S.; Costache, M. Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development—A Focus on Ulcerative Colitis. Cells 2020, 9, 1647. https://doi.org/10.3390/cells9071647
Samoilă I, Dinescu S, Costache M. Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development—A Focus on Ulcerative Colitis. Cells. 2020; 9(7):1647. https://doi.org/10.3390/cells9071647
Chicago/Turabian StyleSamoilă, Iuliana, Sorina Dinescu, and Marieta Costache. 2020. "Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development—A Focus on Ulcerative Colitis" Cells 9, no. 7: 1647. https://doi.org/10.3390/cells9071647
APA StyleSamoilă, I., Dinescu, S., & Costache, M. (2020). Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development—A Focus on Ulcerative Colitis. Cells, 9(7), 1647. https://doi.org/10.3390/cells9071647