CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer

 ,
,  and
and
Abstract
:1. Introduction
2. CSC Subpopulation
2.1. Biological Characteristic of CSCs
2.2. CSC Plasticity
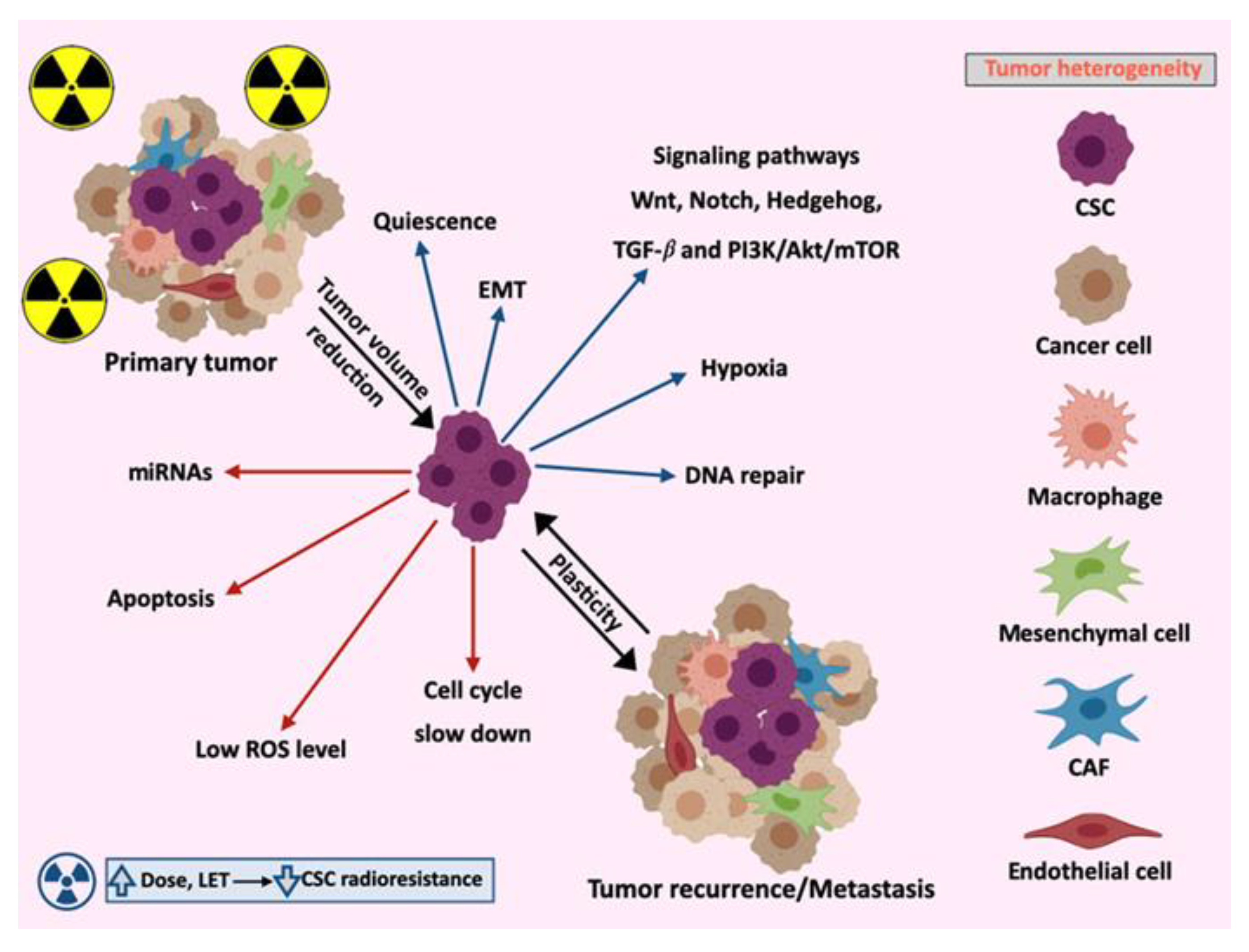
2.3. Tumor Heterogeneity
3. Molecular Mechanisms Involved in CSCs Radioresistance
3.1. Signaling Pathways
3.1.1. Wnt/β-catenin
3.1.2. Notch
3.1.3. Hedgehog (Hh)
3.1.4. TGF-β
3.1.5. PI3K/AKT/mTOR
3.2. Apoptosis
3.3. Cell Cycle
3.4. Epithelial–Mesenchymal Transition (EMT)
3.5. MicroRNAs
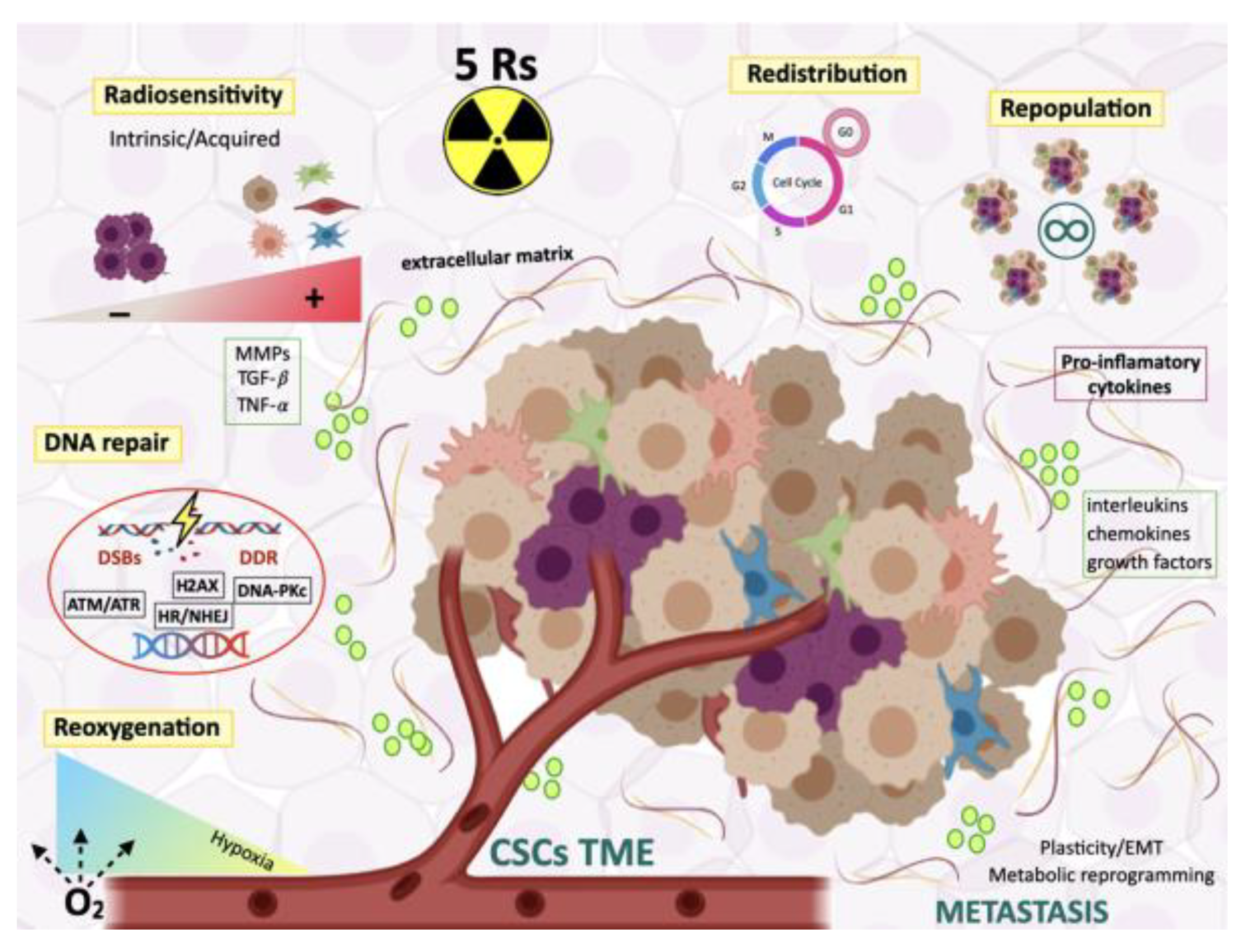
4. CSCs and Microenvironment
4.1. Crosstalk between CSCs and Their Niches
4.2. CSCs Niches in the Primary Tumor and Metastasis
4.3. CSCs and Microenvironment in Response to Radiation
5. Radiocurability and Radiation Therapy Resistance
5.1. The Five Rs of Radiotherapy
5.1.1. Radiosensitivity
5.1.2. Redistribution Through the Cell Cycle
5.1.3. Repopulation of Surviving Normal and Malignant Cells Between Dose Fractions
5.1.4. Repair of Radioinduced DNA Damage
5.1.5. Reoxygenation
5.2. CSCs: Targets for Radiosensitization
5.3. Therapeutic Targeting of CSCs Metabolism
5.4. Targeting Redox Homeostasis
6. Future Perspectives in Radiation Oncology Targeting CSCs: Novel Treatment Approaches
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette | 18F-FLT | 18F-fluorothymidine | NSCLC | non-small cell lung carcinoma |
| Akt | protein kinase B | FGF | fibroblast growth factor | Oct-4 | octamer binding protein 4 |
| ALDH | enzyme aldehyde dehydrogenase | FOXO | forkhead Box class O | OER | oxygen enhancement ratio |
| APLN | angiopoietins, ephrins, apelin | γ-H2AX | phosphorylated Histone 2AX | PDGF | platelet-derived growth factor |
| ATM | ataxia-telangiectasia mutated | GS | γ secretase | PET | positron emission tomography |
| ATR | ataxia telangiectasia and Rad3-related protein | GSIs | γ-secretase inhibitors | PlGF | placental growth factor |
| Bcl-2 | B-cell lymphoma 2 | GSK-3β | glycogen synthase kinase 3 beta | PTEN | phosphatase and tensin homolog |
| BCSC | breast cancer stem cells | HDAC | histone deacetylases | PI3K | phosphatidylinositol 3-kinase |
| BSO | buthionine ulfoximine | HGF | hepatocyte growth factor | RAD51 | |
| CAFs | cancer-associated fibroblasts | Hh | hedgehog signaling | ROS | reactive oxygen species |
| CAIX | carbonic anhydrase IX | HIF1a | hypoxia-inducible factor-1a | RBE | relative biological effectiveness |
| CDC20 | cell-division cycle protein 20 | HR | homologous repair | ROS | reactive oxygen species |
| CD133 | prominin I | ICB | immune checkpoint blockade | RT | radiotherapy |
| CHART | continuous hyperfractionated accelerated radiotherapy trial | ICIs | immune checkpoint inhibitors | SBRT | stereotactic body radiation therapy |
| chks | checkpoints | IL1β | interleukine 1β | SSEA | stage-specific embryonic antigen |
| CHK1 | checkpoint kinase 1 | IL4 | interleukine 4 | SP | side population |
| CHK2 | checkpoint kinase 2 | IL6 | interleukine 6 | TAMs | tumor-associated macrophages |
| CSC | cancer stem cells | IL8 | interleukine 8 | TANs | tumor-associated neutrophils |
| CTC | circulating tumor cells | IL10 | interleukine 10 | TIMPs | endogenous metalloproteinase inhibitors |
| CXC | C-X-C motif chemokines | LET | linear energy transfer | TGF-ß | transforming growth factor-ß |
| CXCL12 C-X-C | motif chemokine ligand 12 | M1 | classically activated macrophage | TGF-ß1 | transforming growth factor-ß1 |
| DAHANCA | Danish Head and Neck Cancer trial | M2 | alternatively activated macrophage | TNF-α | tumor necrosis factor-alpha |
| DAMPs | damage associated molecular patterns | MDSC | myeloid-derived suppressor cells | TICs | tumor initiating cells |
| DCA | dichloroacetate | MiRNAs | microRNAs | TME | tumor microenvironment |
| DDR | DNA damage response | MMPs | matrix metalloproteases | TRAIL | Fas receptor and TNF-related apoptosis inducing ligand |
| DNA | deoxyribonucleic Acid | mRNA | messenger RNA | Treg | regulatory T cells |
| DNA-PKc | DNA-dependent protein kinase, catalytic subunit | mTOR | mammalian target of rapamycin | TSCs | tumor stem cells |
| DSBs | double-strand breaks | NFκB | nuclear factor-κB | UPR | unfolded protein response |
| ECM | extracellular matrix | NHEJ | non-homologous end joining | UV | ultraviolet |
| EGF | epidermal growth factor | NRF2 | nuclear factor-erythroid 2 p45-related factor 2 | VEGF | vascular endothelial growth factor |
| EMT | epithelial-mesenchymal transition | NOD/SCID | nonobese diabetic/severe combined immunodeficient | Wnt | wingless/integrated |
References
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 2013, 108, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad Sci. USA 2007, 104, 618–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Blazek, E.R.; Foutch, J.L.; Maki, G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133− cells, and the CD133+ sector is enlarged by hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Hsu, C.C.; Yung, M.C.; Chen, K.Y.; Tzao, C.; Wu, W.F.; Chou, H.Y.; Lee, Y.Y.; Lu, K.H.; Chiou, S.H.; et al. Enhanced radiosensitivity and radiation-induced apoptosis in glioma CD133-positive cells by knockdown of SirT1 expression. Biochem. Biophys. Res. Commun. 2009, 380, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Chen, Y.W.; Tsai, P.H.; Tsai, M.L.; Lee, Y.Y.; Chiang, C.Y.; Kao, C.L.; Chiou, S.H.; Ku, H.H.; Lin, C.H.; et al. Evaluation of radiotherapy effect in resveratrol-treated medulloblastoma cancer stem-like cells. Childs Nerv. Syst. 2009, 25, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Maverakis, E.; Cornelius, L.A.; Bowen, G.M.; Phan, T.; Patel, F.B.; Fitzmaurice, S.; He, Y.; Burrall, B.; Duong, C.; Kloxin, A.M.; et al. Metastatic melanoma—A review of current and future treatment options. Acta Derm. Venereol. 2015, 95, 516–524. [Google Scholar] [CrossRef] [Green Version]
- West, C.M.; Davidson, S.E.; Elyan, S.A.; Swindell, R.; Roberts, S.A.; Orton, C.J.; Coyle, C.A.; Valentine, H.; Wilks, D.P.; Hunter, R.D.; et al. The intrinsic radiosensitivity of normal and tumour cells. Int. J. Radiat. Biol. 1998, 73, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Balmukhanov, S.B.; Yefimov, M.L.; Kleinbock, T.S. Acquired radioresistance of tumour cells. Nature 1967, 216, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Bratman, S.V.; Diehn, M. Overcoming radioresistance of lung cancer stem cells. In Stem Cells and Cancer Stem Cells: Therapeutic Applications in Disease and Injury; Hayat, M.A., Ed.; Springer: Dordrecht, The Netherlands, 2014; Volume 12, pp. 117–127. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Wagner, V.P.; Martins, M.A.; Martins, M.D.; Warner, K.A.; Webber, L.P.; Squarize, C.H.; Nör, J.E.; Castilho, R.M. Overcoming adaptive resistance in mucoepidermoid carcinoma through inhibition of the IKK-β/IκBα/NFκB axis. Oncotarget 2016, 7, 73032–73044. [Google Scholar] [CrossRef] [Green Version]
- Bighetti-Trevisan, R.L.; Sousa, L.O.; Castilho, R.M.; Almeida, L.O. Cancer stem cells: Powerful targets to improve current anticancer therapeutics. Stem. Cells Int. 2019, 2019, 9618065. [Google Scholar] [CrossRef]
- Ohnishi, H.; Shirato, H.; Nagata, Y.; Hiraolka, M.; Fujino, M.; Gomi, K.; Niibe, Y.; Karasawa, K.; Hayakawa, K.; Takai, Y.; et al. Hypofractinated stereoptactic radiotherapy (HypoFXSRT) for stage I non-small cell lung cancer: Updated results of 257 patients in a Japnese multi-institutional study. J. Thorac. Oncol. 2007, 2 (Suppl. 3), S94–S100. [Google Scholar] [CrossRef] [Green Version]
- Sanuki, N.; Takeda, A.; Oku, Y.; Mizuno, T.; Aoki, Y.; Eriguchi, T.; Iwabuchi, S.; Kunieda, E. Stereotactic body radiotherapy for small hepatocellular carcinoma: A retrospective outcome analysis in 185 patients. Acta Oncol. 2014, 53, 399–404. [Google Scholar] [CrossRef]
- Freeman, D.E.; King, C.R. Stereotactic body radiotherapy for low risk prostate cancer: Five-year outcomes. Radio Oncol. 2011, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioyama, Y.; Onishi, H.; Takayama, K.; Matsuo, Y.; Takeda, A.; Yamashita, H.; Miyakawa, A.; Murakami, N.; Aoki, M.; Matsushita, H.; et al. Clinical outcomes of stereotactic body radiotherapy for patients with stage I small-cell lung cancer: Analysis of a subset of the Japanese radiological society multi-institutional SBRT study group database. Technol. Cancer Res. Treat. 2018, 17, 1533033818783904. [Google Scholar] [CrossRef] [PubMed]
- Goethals, P.M.; Zimmermann, R. Proton Therapy. World Market Report & Drectory, 6th ed.; MEDraysintell: Louvain-la-Neuve, Belgium, 2018; pp. 226–279. [Google Scholar]
- Al-Hajj, M.; Clarke, M.F. Self-renewal and solid tumor stem cells. Oncogene 2004, 23, 7274–7282. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, S. Cancer, stem cells and cancer stem cells: Old ideas, new developments. F1000 Med. Rep. 2011, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Palomeras, S.; Ruiz-Martínez, S.; Puig, T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules 2018, 23, 2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Evers, P.; Lee, P.P.; De Marco, J.; Agazaryan, N.; Sayre, J.W.; Selch, M.; Pajonk, F. Irradiation of the potential cancer stem cell niches in the adult brain improves progression-free survival of patients with malignant glioma. BMC Cancer 2010, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zöller, M. Exosomes, metastases, and the miracle of cancer stem cell markers. Cancer Metastasis Rev. 2019, 38, 259–295. [Google Scholar] [CrossRef]
- Vermeulen, L.; de Sousa e Melo, F.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol. 2012, 13, e83–e89. [Google Scholar] [CrossRef]
- Ablett, M.P.; Singh, J.K.; Clarke, R.B. Stem cells in breast tumours: Are they ready for the clinic? Eur. J. Cancer 2012, 48, 2104–2116. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The role of microRNAs in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Dong, Q.; Li, J.; Zhang, K.; Qin, J.; Zhao, J.; Sun, Q.; Wang, Z.; Wartmann, T.; Jauch, K.W.; et al. Targeting cancer stem cells and their niche: Perspectives for future therapeutic targets and strategies. Semin. Cancer Biol. 2018, 53, 139–155. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Hernández-Camarero, P.; Jiménez, G.; López-Ruiz, E.; Barungi, S.; Marchal, J.A.; Perán, M. Revisiting the dynamic cancer stem cell model: Importance of tumour edges. Crit. Rev. Oncol. Hematol. 2018, 131, 35–45. [Google Scholar] [CrossRef]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef]
- Huang, R.; Zong, X. Aberrant cancer metabolism in epithelial–mesenchymal transition and cancer metastasis: Mechanisms in cancer progression. Crit. Rev. Oncol. Hematol. 2017, 115, 13–22. [Google Scholar] [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nisticò, P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Jiménez, G.; Hackenberg, M.; Catalina, P.; Boulaiz, H.; Griñán-Lisón, C.; García, M.Á.; Perán, M.; López-Ruiz, E.; Ramírez, A.; Morata-Tarifa, C.; et al. Mesenchymal stem cell’s secretome promotes selective enrichment of cancer stem-like cells with specific cytogenetic profile. Cancer Lett. 2018, 429, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, Q.; Shanti, R.M.; Shi, S.; Chang, T.H.; Carrasco, L.; Alawi, F.; Le, A.D. Mesenchymal stromal cell-derived Interleukin-6 promotes epithelial-mesenchymal transition and acquisition of epithelial stem-like cell properties in ameloblastoma epithelial cells. Stem Cells 2017, 35, 2083–2094. [Google Scholar] [CrossRef] [Green Version]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picon-Ruiz, M.; Pan, C.; Drews-Elger, K.; Jang, K.; Besser, A.H.; Zhao, D.; Morata-Tarifa, C.; Kim, M.; Ince, T.A.; Azzam, D.J.; et al. Interactions between adipocytes and breast cancer cells stimulate cytokine production and drive Src/Sox2/miR-302b-mediated malignant progression. Cancer Res. 2016, 76, 491–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Alfonso, J.C.L.; Berk, L. Modeling the effect of intratumoral heterogeneity of radiosensitivity on tumor response over the course of fractionated radiation therapy. Radiat. Oncol. 2019, 14, 88. [Google Scholar] [CrossRef] [Green Version]
- Pribluda, A.; de la Cruz, C.C.; Jackson, E.L. Intratumoral heterogeneity: From diversity comes resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [Green Version]
- Hausser, J.; Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 2020, 20, 247–257. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise review: Cancer cells, cancer stem cells, and mesenchymal stem cells: Influence in cancer development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, M.H.; Kim, K.S.; Park, M.J.; Jeong, J.H.; Park, S.W.; Ji, Y.H.; Kim, K.I.; Lee, T.S.; Ryu, P.Y.; et al. In vivo monitoring of CD44+ cancer stem-like cells by γ-irradiation in breast cancer. Int. J. Oncol. 2016, 48, 2277–2286. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.S.; Hur, W.; Kim, T.K.; Hong, S.W.; Kim, S.W.; Choi, J.E.; Sung, P.S.; Song, M.J.; Lee, B.C.; Hwang, D.; et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012, 315, 129–137. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tao, L.; Yi, J.; Song, H.; Chen, L. The role of canonical Wnt signaling in regulating radioresistance. Cell Physiol. Biochem. 2018, 48, 419–432. [Google Scholar] [CrossRef]
- Luo, M.; Wu, C.; Guo, E.; Peng, S.; Zhang, L.; Sun, W.; Liu, D.; Hu, G.; Hu, G. FOXO3a knockdown promotes radioresistance in nasopharyngeal carcinoma by inducing epithelial-mesenchymal transition and the Wnt/β-catenin signaling pathway. Cancer Lett. 2019, 455, 26–35. [Google Scholar] [CrossRef]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yang, G.; Ding, Y.; Dai, Y.; Xu, S.; Guo, Q.; Xie, A.; Hu, G. Inhibition of PI3K/AKT signaling pathway radiosensitizes pancreatic cancer cells with ARID1A deficiency in vitro. J. Cancer 2018, 9, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.C.; Hung, S.K.; Lin, H.Y.; Chiou, W.Y.; Lee, M.S.; Liao, H.F.; Huang, H.B.; Ho, H.C.; Su, Y.C. Targeting the PI3K/AKT/mTOR signaling pathway as an effectively radiosensitizing strategy for treating human oral squamous cell carcinoma in vitro and in vivo. Oncotarget 2017, 8, 68641–68653. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Czochor, J.R.; Glazer, P.M. MicroRNAs in cancer cell response to ionizing radiation. Antioxid. Redox. Signal 2014, 21, 293–312. [Google Scholar] [CrossRef]
- Jan, R.; Gul-e-Saba, C. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y.J. Cell death response to dna damage. Yale J. Biol. Med. 2019, 92, 771–779. [Google Scholar] [PubMed]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.J.; Vance, L.D.; Psioda, M.; Smith-Roe, S.L.; Simpson, D.A.; Ibrahim, J.G.; Hoadley, K.A.; Perou, C.M.; Kaufmann, W.K. Patterns of cell cycle checkpoint deregulation associated with intrinsic molecular subtypes of human breast cancer cells. NPJ Breast Cancer 2017, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.X.; Poovey, C.E.; Privette, A.A.; Grant, G.D.; Chao, H.Y.; Cook, J.G.; Purvis, J.E. Orchestration of DNA damage checkpoint dynamics across the human cell cycle. Cell Syst. 2017, 5, 445e.5–459.e5. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef]
- Rezaeian, A.H.; Khanbabaei, H.; Calin, G.A. Therapeutic potential of the miRNA–ATM axis in the management of tumor radioresistance. Cancer Res. 2020, 80, 139–150. [Google Scholar] [CrossRef]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.I.; Ganswindt, U. The role of cancer stem cells in radiation resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Von Essen, C.F. Radiation enhancement of metastasis: A review. Clin. Exp. Metastasis 1991, 9, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Madani, I.; De Neve, W.; Mareel, M. Does ionizing radiation stimulate cancer invasion and metastasis? Bull. Cancer 2008, 95, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Jang, S.J.; Kang, S.W.; Park, S.; Hwang, S.G.; Kim, W.J.; Kang, J.H.; Um, H.D. Establishment of animal model for the analysis of cancer cell metastasis during radiotherapy. Radiat. Oncol. 2012, 7, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncharmont, C.; Levy, A.; Guy, J.B.; Falk, A.T.; Guilbert, M.; Trone, J.C.; Alphonse, G.; Gilormini, M.; Ardail, D.; Toillon, R.A.; et al. Radiation-enhanced cell migration/invasion process: A review. Crit. Rev. Oncol. Hematol. 2014, 92, 133–142. [Google Scholar] [CrossRef]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar]
- Wang, H.; Wang, Z.; Li, Y.; Lu, T.; Hu, G. Silencing snail reverses epithelial-mesenchymal transition and increases radiosensitivity in hypopharyngeal carcinoma. Onco. Targets Ther. 2020, 13, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Kim, A.R.; Nam, J.K.; Kim, J.M.; Kim, J.Y.; Seo, H.R.; Lee, H.J.; Cho, J.; Lee, Y.J. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6+ cancer cell and macrophage polarization. Nat. Commun. 2018, 9, 5108. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Zhang, X.; Bai, J.; Li, Y.; Wang, X.; Zhou, Y. Tissue-specific and exosomal miRNAs in lung cancer radiotherapy: From regulatory mechanisms to clinical implications. Cancer Manag. Res. 2019, 11, 4413–4424. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Sultana, M.; Qazi, A.; Qazi, M.H.; Parveen, G.; Waquar, S.; Ashraf, A.B.; Rasool, M. Role of natural radiosensitizers and cancer cell radioresistance: An update. Anal. Cell. Pathol. 2016, 2016, 6146595. [Google Scholar] [CrossRef] [Green Version]
- Dudás, J.; Ladányi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to mesenchymal transition: A mechanism that fuels cancer radio/chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef] [Green Version]
- Metheetrairut, C.; Slack, F.J. MicroRNAs in the ionizing radiation response and in radiotherapy. Curr. Opin. Genet. Dev. 2013, 23, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dando, I.; Cordani, M.; Pozza, E.D.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant mechanisms and ROS-related MicroRNAs in cancer stem cells. Oxid. Med. Cell. Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef] [Green Version]
- Griñán-Lisón, C.; Olivares-Urbano, M.A.; Jiménez, G.; López-Ruiz, E.; del Val, C.; Morata-Tarifa, C.; Entrena, J.M.; González-Ramírez, A.R.; Boulaiz, H.; Zurita Herrera, M.; et al. miRNAs as radio-response biomarkers for breast cancer stem cells. Mol. Oncol. 2020, 14, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Urbano, M.A.; Griñán-Lisón, C.; Ríos-Arrabal, S.; Artacho-Cordón, F.; Torralbo, A.I.; López-Ruiz, E.; Marchal, J.A.; Núñez, M.I. Radiation and stemness phenotype may influence individual breast cancer outcomes: The crucial role of MMPs and microenvironment. Cancers 2019, 11, 1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, S.K.; Guan, J.L. Breast cancer: Multiple subtypes within a tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef]
- Mondal, S.; Bhattacharya, K.; Mandal, C. Nutritional stress reprograms dedifferention in glioblastoma multiforme driven by PTEN/Wnt/Hedgehog axis: A stochastic model of cancer stem cells. Cell Death Discov. 2018, 4, 110. [Google Scholar] [CrossRef] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Atiya, H.; Frisbie, L.; Pressimone, C.; Coffman, L. Mesenchymal stem cells in the tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 31–42. [Google Scholar] [CrossRef]
- Schaue, D.; Micewicz, E.D.; Ratikan, J.A.; Xie, M.W.; Cheng, G.; McBride, W.H. Radiation and inflammation. Semin. Radiat. Oncol. 2015, 25, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, F.; Tsai, Y.; Yang, X.; Yang, L.; Duan, S.; Wang, X.; Keng, P.; Lee, S.O. IL-6 signaling promotes DNA repair and prevents apoptosis in CD133+ stem-like cells of lung cancer after radiation. Radiat. Oncol. 2015, 10, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Su, Y.; Zhu, H.; Wang, X.; Li, X.; Dai, C.; Xu, C.; Zheng, T.; Mao, C.; Chen, D. Interleukin-23 receptor signaling mediates cancer dormancy and radioresistance in human esophageal squamous carcinoma cells via the Wnt/Notch pathway. J. Mol. Med. 2019, 97, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic Hedgehog signaling in organogenesis, tumors, and tumor microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [Green Version]
- Koledova, Z. 3D cell culture: An introduction. In 3D Cell Culture: Methods and Protocols; Koledova, Z., Ed.; Humana Press: New York, NY, USA, 2017; pp. 1–11. [Google Scholar] [CrossRef]
- Sattiraju, A.; Sai, K.K.S.; Mintz, A. Glioblastoma stem cells and their microenvironment. Adv. Exp. Med. Biol. 2017, 1041, 119–140. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Ingangi, V.; Minopoli, M.; Ragone, C.; Motti, M.L.; Carriero, M.V. Role of microenvironment on the fate of disseminating cancer stem cells. Front. Oncol. 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Das, P.K.; Pillai, S.; Rakib, M.A.; Khanam, J.A.; Gopalan, V.; Lam, A.K.Y.; Islam, F. Plasticity of cancer stem cell: Origin and role in disease progression and therapy resistance. Stem Cell Rev. Rep. 2020, 16, 397–412. [Google Scholar] [CrossRef]
- Ahmed, F.; Haass, N.K. Microenvironment-driven dynamic heterogeneity and phenotypic plasticity as a mechanism of melanoma therapy resistance. Front. Oncol. 2018, 8, 173. [Google Scholar] [CrossRef]
- Davies, A.E.; Albeck, J.G. Microenvironmental signals and biochemical information processing: Cooperative determinants of intratumoral plasticity and heterogeneity. Front. Cell Dev. Biol. 2018, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Nascimento-Filho, C.H.V.; Webber, L.P.; Borgato, G.B.; Goloni-Bertollo, E.M.; Squarize, C.H.; Castilho, R.M. Hypoxic niches are endowed with a protumorigenic mechanism that supersedes the protective function of PTEN. FASEB J. 2019, 33, 13435–13449. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Artacho-Cordón, A.; Artacho-Cordón, F.; Ríos-Arrabal, S.; Calvente, I.; Núñez, M.I. Tumor microenvironment and breast cancer progression: A complex scenario. Cancer Biol. Ther. 2012, 13, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artacho-Cordón, F.; Ríos-Arrabal, S.; Lara, P.C.; Artacho-Cordón, A.; Calventea, I.; Núñez, M.I. Matrix metalloproteinases: Potential therapy to prevent the development of second malignancies after breast radiotherapy. Surg. Oncol. 2012, 21, e143–e151. [Google Scholar] [CrossRef]
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.K.K.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 6838. [Google Scholar] [CrossRef]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; You, M.J.; Yang, Y.; Hu, D.; Tian, C. The role of tumor-associated macrophages in leukemia. Acta Haemato. 2020, 143, 112–117. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Kamińska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Roles of long noncoding RNAs in recurrence and metastasis of radiotherapy-resistant cancer stem cells. Int. J. Mol. Sci. 2017, 18, 1903. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichon, T.; Kulach, N. Tumor microenvironment as a “game changer” in cancer radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.S.; Hahn, S.M. Immune priming of the tumor microenvironment by radiation. Trends Cancer 2016, 2, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Miao, L.; St Clair, D.K.; St Clair, W.H. Redox-modulated phenomena and radiation therapy: The central role of superoxide dismutases. Antioxid. Redox. Signal. 2014, 20, 1567–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuonen, F.; Secondini, C.; Rüegg, C. Molecular pathways: Emerging pathways mediating growth, invasion, and metastasis of tumors progressing in an irradiated microenvironment. Clin. Cancer Res. 2012, 18, 5196–5202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofia Vala, I.; Martins, L.R.; Imaizumi, N.; Nunes, R.J.; Rino, J.; Kuonen, F.; Carvalho, L.M.; Rüegg, C.; Grillo, I.M.; Barata, J.T.; et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS ONE 2010, 5, e11222. [Google Scholar] [CrossRef]
- Chung, Y.L.; Jian, J.J.; Cheng, S.H.; Tsai, S.Y.; Chuang, V.P.; Soong, T.; Lin, Y.M.; Horng, C.F. Sublethal irradiation induces vascular endothelial growth factor and promotes growth of hepatoma cells: Implications for radiotherapy of hepatocellular carcinoma. Clin. Cancer Res. 2006, 12, 2706–2715. [Google Scholar] [CrossRef] [Green Version]
- Kurth, I.; Peitzsch, C.; Baumann, M.; Dubrovska, A. The role of cancer stem cells in tumor radioresistance. In Cancer Stem Cells; Rajasekhar, V.K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 473–491. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.K. HIF-1 signaling: A key orchestrator of cancer radioresistance. Radiat. Med. Prot. 2020, 1, 7–14. [Google Scholar] [CrossRef]
- Elming, P.B.; Sorensen, B.S.; Oei, A.L.; Franken, N.A.P.; Crezee, J.; Overgaard, J.; Horsman, M.R. Hyperthermia: The optimal treatment to overcome radiation resistant hypoxia. Cancers 2019, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- Baumann, R.; Depping, R.; Delaperriere, M.; Dunst, J. Targeting hypoxia to overcome radiation resistance in head & neck cancers: Real challenge or clinical fairytale? Expert Rev. Anticancer Ther. 2016, 16, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Epel, B.; Maggio, M.C.; Barth, E.D.; Miller, R.C.; Pelizzari, C.A.; Krzykawska-Serda, M.; Sundramoorthy, S.V.; Aydogan, B.; Weichselbaum, R.R.; Tormyshev, V.M.; et al. Oxygen-Guided radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z. Increased oxidative stress as a selective anticancer therapy. Oxid. Med. Cell Longev. 2015, 2015, 294303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoning, J.P.; Monteiro, M.; Gu, W. Drug resistance and cancer stem cells: The shared but distinct roles of hypoxia-inducible factors HIF1α and HIF2α. Clin. Exp. Pharmacol. Physiol. 2017, 44, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Takam Kamga, P.; Tagne Simo, R.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Kumar Goel, R.; Mbo Amvene, J.; et al. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol. Med. 2017, 14, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Sil, P.C. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol. Res. 2019, 142, 251–261. [Google Scholar] [CrossRef]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Cojoc, M.; Peitzsch, C.; Kurth, I.; Trautmann, F.; Kunz-Schughart, L.A.; Telegeev, G.D.; Stakhovsky, E.A.; Walker, J.R.; Simin, K.; Lyle, S.; et al. Aldehyde dehydrogenase is regulated by β-Catenin/TCF and promotes radioresistance in prostate cancer progenitor cells. Cancer Res. 2015, 75, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.T.; Lin, W.Y.; Chang, Y.H.; Chen, W.C.; Chen, M.F. Impact of CD44 expression on radiation response for bladder cancer. J. Cancer 2017, 8, 1137–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins-Neves, S.R.; Cleton-Jansen, A.M.; Gomes, C.M.F. Therapy-induced enrichment of cancer stem-like cells in solid human tumors: Where do we stand? Pharmacol. Res. 2018, 137, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Thames, H.; Trott, K.; Zips, D. Cancer stem cells and radiotherapy. Int. J. Radiat. Biol. 2009, 85, 391–402. [Google Scholar] [CrossRef]
- Yaromina, A.; Krause, M.; Thames, H.; Rosner, A.; Krause, M.; Hessel, F.; Grenman, R.; Zips, D.; Baumann, M. Pre-treatment number of clonogenic cells and their radiosensitivity are major determinants of local tumour control after fractionated irradiation. Radiother. Oncol. 2007, 83, 304–310. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef]
- Ogawa, K.; Boucher, Y.; Kashiwagi, S.; Fukumura, D.; Chen, D.; Gerweck, L.E. Influence of tumor cell and stroma sensitivity on tumor response to radiation. Cancer Res. 2007, 67, 4016–4021. [Google Scholar] [CrossRef] [Green Version]
- Dubben, H.H.; Thames, H.D.; Beck-Bornholdt, H.P. Tumor volume: A basic and specific response predictor in radiotherapy. Radiother. Oncol. 1998, 47, 167–174. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Zaidi, S.T.; Zietman, A. Multivariate determinants of radiocurability. I: Prediction of single fraction tumor control doses. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 57–66. [Google Scholar] [CrossRef]
- Steel, G.G.; McMillan, T.J.; Peacock, J.H. The 5 Rs of radiobiology. Int. J. Radiat. Biol. 1989, 56, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi, H.; Shiri, I.; Atashzar, M.; Sarebani, M.; Moloudi, K.; Samadian, H. Radiation protection and secondary cancer prevention using biological radioprotectors in radiotherapy. Int. J. Cancer Ther. Oncol. 2015, 3, 335. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M. CD44: A cancer stem cell-related biomarker with predictive potential for radiotherapy. Clin. Cancer Res. 2010, 16, 5091–5093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, W.K.; Morton, R.A. X-Ray and ultraviolet sensitivity of symchronized chinese hámster cells at various stages of the cell cycle. Biophys. J. 1965, 5, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Withers, H.R. Cell cycle redistribution as a factor in multifraction irradiation. Radiology 1975, 114, 199–202. [Google Scholar] [CrossRef]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; di Fiore, P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010, 140, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ma, Z.; Xiao, Z.; Liu, H.; Dou, Z.; Feng, X.; Shi, H. Chk1 knockdown confers radiosensitization in prostate cancer stem cells. Oncol. Rep. 2012, 28, 2247–2254. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Glass, J. The phenotypic radiation resistance of CD44+/CD24(-or low) breast cancer cells is mediated through the enhanced activation of ATM signalling. PLoS ONE 2011, 6, e24080. [Google Scholar] [CrossRef]
- Hirose, H.; Ishii, H.; Mimori, K.; Ohta, D.; Ohkuma, M.; Tsujii, H.; Saito, T.; Sekimoto, M.; Doki, Y.; Mori, M. Notch pathway as candidate therapeutic target in Her2/Neu/ErbB2 receptor-negative breast tumors. Oncol. Rep. 2010, 23, 35–43. [Google Scholar]
- Vlashi, E.; Kim, K.; Lagadec, C.; Donna, L.D.; McDonald, J.T.; Eghbali, M.; Sayre, J.W.; Stefani, E.; McBride, W.; Pajonk, F. In-vivo imaging, tracking, and targeting of cancer stem cells. J. Natl. Cancer Inst. 2009, 101, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Bese, N.S.; Sut, P.A.; Ober, A. The effect of treatment interruptions in the postoperative irradiation of breast cancer. Oncology 2005, 69, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Hur, B.I.; Ko, M.H.; Kim, C.H.; Cha, S.H.; Kang, S.K. Potential identity of multi-potential cancer stem-like subpopulation after radiation of cultured brain glioma. BMC Neurosci. 2008, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004, 75, 355–373. [Google Scholar]
- Diehn, M.; Clarke, M.F. Cancer stem cells and radiotherapy: New insights into tumor radioresistance. J. Natl. Cancer Inst. 2006, 98, 1755–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Kantake, N.; Sugiyama, T.; Kowalczykowski, S.C. Rad51 protein controls Rad52-mediated DNA annealing. J. Biol. Chem. 2008, 283, 14883–14892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Nordsmark, M.; Bentzen, S.M.; Rudat, V.; Brizel, D.; Lartigau, E.; Stadler, P.; Becker, A.; Adam, M.; Molls, M.; Dunst, J.; et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005, 77, 18–24. [Google Scholar] [CrossRef]
- Sato, K.; Shimokawa, T.; Imai, T. Difference in acquired radioresistance induction between repeated photon and particle irradiation. Front. Oncol. 2019, 9, 1213. [Google Scholar] [CrossRef]
- Saga, R.; Matsuya, Y.; Takahashi, R.; Hasegawa, K.; Date, H.; Hosokawa, Y. Analysis of the high-dose-range radioresistance of prostate cancer cells, including cancer stem cells, based on a stochastic model. J. Radiat. Res. 2019, 60, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.N.D.; Mierzwa, M.; D’Silva, N.J. Radiation resistance in head and neck squamous cell carcinoma: Dire need for an appropriate sensitizer. Oncogene 2020, 39, 3638–3649. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Dini, V.; Belli, M.; Tabocchini, M.A. Targeting cancer stem cells: Protons versus photons. Br. J. Radiol. 2019, 93, 20190225. [Google Scholar] [CrossRef]
- Vitti, E.T.; Parsons, J.L. The radiobiological effects of proton beam therapy: Impact on DNA damage and repair. Cancers 2019, 11, 946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, M.; Loeffler, J.S. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 2010, 7, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, S.H.; Fang, B.; Gillin, M.; Mohan, R.; Chang, J.Y. Therapy-resistant cancer stem cells have differing sensitivity to photon versus proton beam radiation. J. Thorac. Oncol. 2013, 8, 1484–1491. [Google Scholar] [CrossRef] [Green Version]
- Narang, H.; Kumar, A.; Bhat, N.; Pandey, B.N.; Ghosh, A. Effect of proton and gamma irradiation on human lung carcinoma cells: Gene expression, cell cycle, cell death, epithelial-mesenchymal transition and cancer-stem cell trait as biological end points. Mutat. Res. 2015, 780, 35–46. [Google Scholar] [CrossRef]
- Pecchia, I.; Dini, V.; Ricci-Vitiani, L.; Biffoni, M.; Balduzzi, M.; Fratini, E.; Belli, M.; Campa, A.; Esposito, G.; Cirrone, G.; et al. Glioblastoma stem cells: Radiobiological response to ionising radiations of different qualities. Radiat. Prot. Dosimetry 2015, 166, 374–378. [Google Scholar] [CrossRef]
- Chiblak, S.; Tang, Z.; Campos, B.; Gal, Z.; Unterberg, A.; Debus, J.; Herold-Mende, C.; Abdollahi, A. Radiosensitivity of patient-derived glioma stem cell 3-dimensional cultures to photon, proton, and carbon irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 112–119. [Google Scholar] [CrossRef]
- Girdhani, S.; Sachs, R.; Hlatky, L. Biological effects of proton radiation: An update. Radiat. Prot. Dosimetry 2015, 166, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, 13961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor cells surviving exposure to proton or photon radiation share a common immunogenic modulation signature, rendering them more sensitive to T cell-mediated killing. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Vares, G.; Jallet, V.; Matsumoto, Y.; Rentier, C.; Takayama, K.; Sasaki, T.; Hayashi, Y.; Kumada, H.; Sugawara, H. Functionalized mesoporous silica nanoparticles for innovative boron-neutron capture therapy of resistant cancers. Nanomedicine 2020, 27, 102195. [Google Scholar] [CrossRef] [PubMed]
- Sai, S.; Suzuki, M.; Kim, E.H.; Hayashi, M.; Vares, G.; Yamamoto, N.; Miyamoto, T. Effects of carbon ion beam alone or in combination with cisplatin on malignant mesothelioma cells in vitro. Oncotarget 2017, 9, 14849–14861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, S.; Wakai, T.; Vares, G.; Yamada, S.; Kamijo, T.; Kamada, T.; Shirai, T. Combination of carbon ion beam and gemcitabine causes irreparable DNA damage and death of radioresistant pancreatic cancer stem-like cells in vitro and in vivo. Oncotarget 2015, 6, 5517–5535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, G.; Maalouf, M.; Boivin, A.; Battiston-Montagne, P.; Beuve, M.; Levy, A.; Jalade, P.; Fournier, C.; Ardail, D.; Magné, N.; et al. Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation. Stem Cell Rev. Rep. 2014, 10, 114–126. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.P.; Chneiweiss, H.; Castera, L.; Müller, E.; Stefan, D.; Saintigny, Y. Radiosensitization effect of talazoparib, a Parp inhibitor, on glioblastoma stem cells exposed to low and high linear energy transfer radiation. Sci. Rep. 2018, 8, 3664. [Google Scholar] [CrossRef]
- Wozny, A.S.; Vares, G.; Alphonse, G.; Lauret, A.; Monini, C.; Magné, N.; Cuerq, C.; Fujimori, A.; Monboisse, J.C.; Beuve, M.; et al. ROS production and distribution: A new paradigm to explain the differential effects of x-ray and carbon ion irradiation on cancer stem cell migration and invasion. Cancers 2019, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Andoh, T.; Fujimoto, T.; Suzuki, M.; Sudo, T.; Sakurai, Y.; Tanaka, H.; Fujita, I.; Fukase, N.; Moritake, H.; Sugimoto, T.; et al. Boron neutron capture therapy (BNCT) as a new approach for clear cell sarcoma (CCS) treatment: Trial using a lung metastasis model of CCS. Appl. Radiat. Isot. 2015, 106, 195–201. [Google Scholar] [CrossRef]
- Futamura, G.; Kawabata, S.; Siba, H.; Kuroiwa, T.; Suzuki, M.; Kondo, N.; Ono, K.; Sakurai, Y.; Tanaka, M.; Todo, T.; et al. A case of radiation-induced osteosarcoma treated effectively by boron neutron capture therapy. Radiat. Oncol. 2014, 9, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedunchezhian, K.; Aswath, N.; Thiruppathy, M.; Thirugnanamurthy, S. Boron neutron capture therapy—A literature review. J. Clin. Diagn. Res. 2016, 10, ZE01–ZE04. [Google Scholar] [CrossRef] [PubMed]
- Jingu, K.; Tsujii, H.; Mizoe, J.E.; Hasegawa, A.; Bessho, H.; Takagi, R.; Morikawa, T.; Tonogi, M.; Tsuji, H.; Kamada, T.; et al. Carbon ion radiation therapy improves the prognosis of unresectable adult bone and soft-tissue sarcoma of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 2125–2131. [Google Scholar] [CrossRef]
- Ishikawa, H.; Tsuji, H.; Kamada, T.; Akakura, K.; Suzuki, H.; Shimazaki, J.; Tsujii, H. Carbon-ion radiation therapy for prostate cancer. Int. J. Urol. 2012, 19, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Ohno, T.; Tsujii, H.; Nakano, T.; Mizoe, J.E.; Kamada, T.; Miyamoto, T.; Tsuji, H.; Kato, H.; Yamada, S.; et al. Dose escalation study of carbon ion radiotherapy for locally advanced carcinoma of the uterine cervix. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Tsujii, H.; Miyamoto, T.; Mizoe, J.E.; Kamada, T.; Tsuji, H.; Yamada, S.; Kandatsu, S.; Yoshikawa, K.; Obata, T.; et al. Results of the first prospective study of carbon ion radiotherapy for hepatocellular carcinoma with liver cirrhosis. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Baba, M.; Sugane, T.; Nakajima, M.; Yashiro, T.; Kagei, K.; Hirasawa, N.; Sugawara, T.; Yamamoto, N.; Koto, M.; et al. Carbon ion radiotherapy for stage I non-small cell lung cancer using a regimen of four fractions during 1 week. J. Thorac. Oncol. 2007, 2, 916–926. [Google Scholar] [CrossRef] [Green Version]
- Mizoe, J.E.; Hasegawa, A.; Jingu, K.; Takagi, R.; Bessyo, H.; Morikawa, T.; Tonoki, M.; Tsuji, H.; Kamada, T.; Tsujii, H.; et al. Results of carbon ion radiotherapy for head and neck cancer. Radiother. Oncol. 2012, 103, 32–37. [Google Scholar] [CrossRef]
- Tinganelli, W.; Durante, M.; Hirayama, R.; Kramer, M.; Maier, A.; Kraft-Weyrather, W.; Furusawa, Y.; Friedrich, T.; Scifoni, E. Kill-painting of hypoxic tumours in charged particle therapy. Sci. Rep. 2015, 5, 17016. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” cancer stem cells (e-CSCs): A new hyper-metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front. Oncol. 2018, 8, 677. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and non tumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Kwon, H.; Han, C.; Zhang, J.; Dash, S.; Lim, K.; Wu, T. Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: Regulation by MIR-122. Oncotarget 2015, 6, 40822–40835. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.A.; Wang, C.Y.; Hsieh, Y.T.; Chen, Y.J.; Wei, Y.H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caria, P.; Tronci, L.; Dettori, T.; Murgia, F.; Santoru, M.L.; Griffin, J.L.; Vanni, R.; Atzori, L. Metabolomic alterations in thyrospheres and adherent parental cells in papillary thyroid carcinoma cell lines: A pilot study. Int. J. Mol. Sci. 2018, 19, 2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; Hulver, M.W.; Schmelz, E.M. Ovarian tumor-initiating cells display a flexible metabolism. Exp. Cell Res. 2014, 328, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Jagust, P.; de Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-based therapeutic strategies targeting cancer stem cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signalling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Yeo, H.; Lyssiotis, C.A.; Zhang, Y.; Ying, H.; Asara, J.M.; Cantley, L.C.; Paik, J.H. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013, 32, 2589–2602. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.W.; Chen, Y.S.; Tsay, Y.G.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Hung, K.F.; Lin, C.H.; Huang, T.Y.; Kao, S.Y.; et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 2018, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Lu, Y.; Yang, J.; Chen, G.; Kim, S.; Feng, L.; Ogasawara, M.; Hammoudi, N.; Lu, W.; Zhang, H.; et al. Metabolic activation of mitochondria in glioma stem cells promotes cancer development through a reactive oxygen species-mediated mechanism. Stem Cell Res. Ther. 2015, 6, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. 2018, 28, 69–86.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, A.; Hanot, M.; Malesys, C.; Maalouf, M.; Rousson, R.; Rodriguez-Lafrasse, C.; Ardail, D. Transient alteration of cellular redox buffering before irradiation triggers apoptosis in head and neck carcinoma stem and non-stem cells. PLoS ONE 2011, 6, e14558. [Google Scholar] [CrossRef] [Green Version]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of radiation response in breast cancer stem cells by inhibition of thioredoxin-and glutathione-dependent metabolism. Radiat. Res. 2016, 186, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Lenz, G.; Batista, L.F.Z. Tumor propagating cells: Drivers of tumor plasticity, heterogeneity, and recurrence. Oncogene 2020, 39, 2055–2068. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.X. Targeting the tumor microenvironment with interferon-β bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozar, S.; Morrissey, E.; Nicholson, A.M.; van der Heijden, M.; Zecchini, H.I.; Kemp, R.; Tavaré, S.; Vermeulen, L.; Winton, D.J. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell 2013, 13, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.; Ellenbroek, S.I.; Ritsma, L.; Beerling, E.; Vrisekoop, N.; Van Rheenen, J. Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells 2013, 31, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisfeld, R.A. The tumor microenvironment: A target for combination therapy of breast cancer. Crit. Rev. Oncog. 2013, 18, 115–133. [Google Scholar] [CrossRef]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT–TCF addiction and enhancement of HH–GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- LaBarge, M.A. The difficulty of targeting cancer stem cell niches. Clin. Cancer Res. 2010, 16, 3121–3129. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Rajguru, S.; Lubner, S.J.; Mulkerin, D.; Schelman, W.R.; Winterle, N.; Leverson, K.D.H.; Chen, H.; LoConte, N.K. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in low-grade neuroendocrine tumors. J. Clin. Oncol. 2012, 30, e14554. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01013597 (accessed on 14 April 2020).
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Beachy, P.A.; Hymowitz, S.G.; Lazarus, R.A.; Leahy, D.J.; Siebold, C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010, 24, 2001–2012. [Google Scholar] [CrossRef] [Green Version]
- Chien, A.J.; Conrad, W.H.; Moon, R.T. A Wnt survival guide: From flies to human disease. J. Invest. Dermatol. 2009, 129, 1614–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzen, S.M.; Atasoy, B.M.; Daley, F.M.; Dische, S.; Richman, P.I.; Saunders, M.I.; Trott, K.R.; Wilson, G.D. Epidermal growth factor receptor expression in pretreatment biopsies from head and neck squamous cell carcinoma as a predictive factor for a benefit from accelerated radiation therapy in a randomized controlled trial. J. Clin. Oncol. 2005, 23, 5560–5567. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, J.G.; Steiniche, T.; Overgaard, J. The influence of epidermal growth factor receptor and tumor differentiation on the response to accelerated radiotherapy of squamous cell carcinomas of the head and neck in the randomized DAHANCA 6 and 7 study. Radiother. Oncol. 2005, 74, 93–100. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Meng, Y.; Dekmezian, C.; Kim, K.; Pajonk, F. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res. 2010, 12, R13. [Google Scholar] [CrossRef] [Green Version]
- Ghisolfi, L.; Keates, A.C.; Hu, X.; Lee, D.K.; Li, C.J. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012, 7, e43628. [Google Scholar] [CrossRef]
- Sato, K.; Nitta, N.; Aoki, I.; Imai, T.; Shimokawa, T. Repeated photon and C-ion irradiations in vivo have different impact on alteration of tumor characteristics. Sci. Rep. 2018, 8, 1458. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.J.; Ishii, H.; Tamari, K.; Hayashi, K.; Nishida, N.; Konno, M.; Kawamoto, K.; Koseki, J.; Fukusumi, T.; Hasegawa, S.; et al. Cancer stem cells: The potential of carbon ion beam radiation and new radiosensitizers (Review). Oncol. Rep. 2015, 34, 2233–2237. [Google Scholar] [CrossRef] [Green Version]
- Kyjacova, L.; Hubackova, S.; Krejcikova, K.; Strauss, R.; Hanzlikova, H.; Dzijak, R.; Imrichova, T.; Simova, J.; Reinis, M.; Bartek, J.; et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, Erk signaling-dependent cells. Cell Death Differ. 2015, 22, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Chen, A.M.; Boyrie, S.; Yu, G.; Nguyen, A.; Brower, P.A.; Hess, C.B.; Pajonk, F. Radiation-induced dedifferentiation of head and neck cancer cells into cancer stem cells depends on human papillomavirus status. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shi, H.; Chen, H.; Gong, A.; Liu, Y.; Song, L.; Xu, X.; You, T.; Fan, X.; Wang, D.; et al. Dedifferentiation process driven by radiotherapy induced HMGB1/TLR2/YAP/HIF-1α signaling enhances pancreatic cancer stemness. Cell Death Dis. 2019, 10, 724. [Google Scholar] [CrossRef] [Green Version]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging opportunities and challenges in cancer immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, S.; Coleman, C.N.; Formenti, S.C. Radiotherapy: Changing the game in immunotherapy. Trends Cancer 2016, 2, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Apporaches to treat immune hot, altered and cold tumors with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Konings, K.; Vandevoorde, C.; Baselet, B.; Baatout, S.; Moreels, M. Combination therapy with charged particles and molecular targeting: A promising avenue to overcome radioresistance. Front. Oncol. 2020, 10, 128. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Identifier | Tumor Type | No. of Patients | Phase/Status | Treatment Schedule | Toxicity/Adverse events (Serious/Not Serious) | Results |
|---|---|---|---|---|---|---|
| NCT01868503 | Locally advanced or locally recurrent breast cancer that is refractory to chemotherapy | 7 | II/Terminated (protocol modification) | Conventional RT + Lapatinib ditosylate | Lymphocytes count decreased/anemia/ fever-possible sepsis/endocrine disorders | Change in the proportion of BCSCs not analyzed |
| NCT02039778 | Brain tumor | 4 | Not applicable/ Terminated (poor accrual) | Stem cell RT/IMRT + Temozolamide | Death†/platelet count decreased†/blurred vision†/fatigue†/ nausea†/ headache†/dry skin† | Not completed |
| NCT04031378 | Oligometastatic prostate | 100 | II/Not yet recruiting | Single Dose RT (SDRT) with or without adjuvant systemic therapy | Not provided | No results posted |
| NCT03085992 | Resectable rectal cancer | 49 | II/Completed | FOLFOXIRI † Bevacizumab Chemoradiotherapy † Bevacizumab | Not provided | No results posted |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivares-Urbano, M.A.; Griñán-Lisón, C.; Marchal, J.A.; Núñez, M.I. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells 2020, 9, 1651. https://doi.org/10.3390/cells9071651
Olivares-Urbano MA, Griñán-Lisón C, Marchal JA, Núñez MI. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells. 2020; 9(7):1651. https://doi.org/10.3390/cells9071651
Chicago/Turabian StyleOlivares-Urbano, María Auxiliadora, Carmen Griñán-Lisón, Juan Antonio Marchal, and María Isabel Núñez. 2020. "CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer" Cells 9, no. 7: 1651. https://doi.org/10.3390/cells9071651
APA StyleOlivares-Urbano, M. A., Griñán-Lisón, C., Marchal, J. A., & Núñez, M. I. (2020). CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells, 9(7), 1651. https://doi.org/10.3390/cells9071651