Role of Mitochondria in Cancer Stem Cell Resistance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondria as a Crucial Element in Cancer
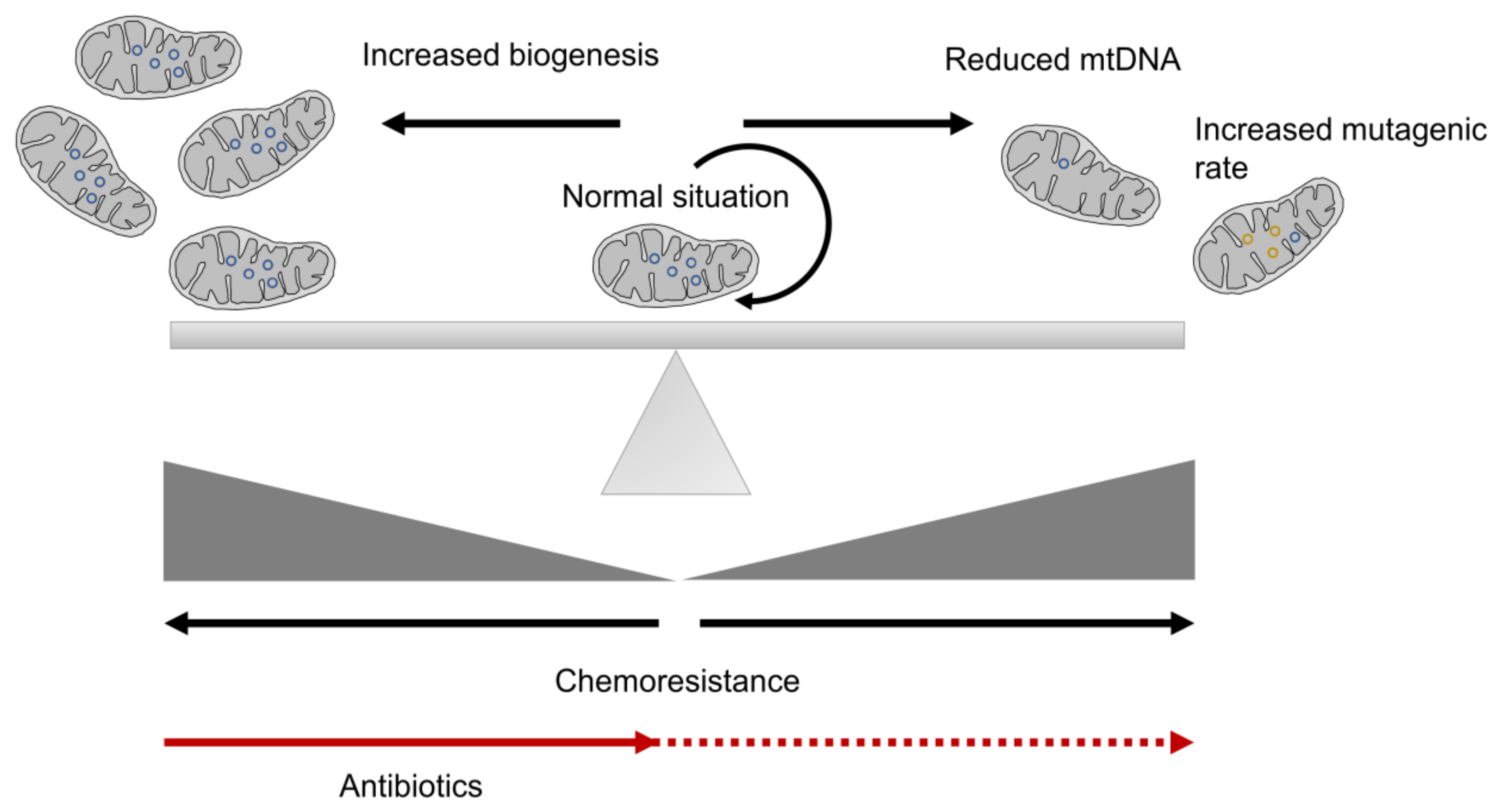
3. Mitochondrial Biogenesis and CSC Resistance
4. Mitochondrial Metabolism in CSC Resistance
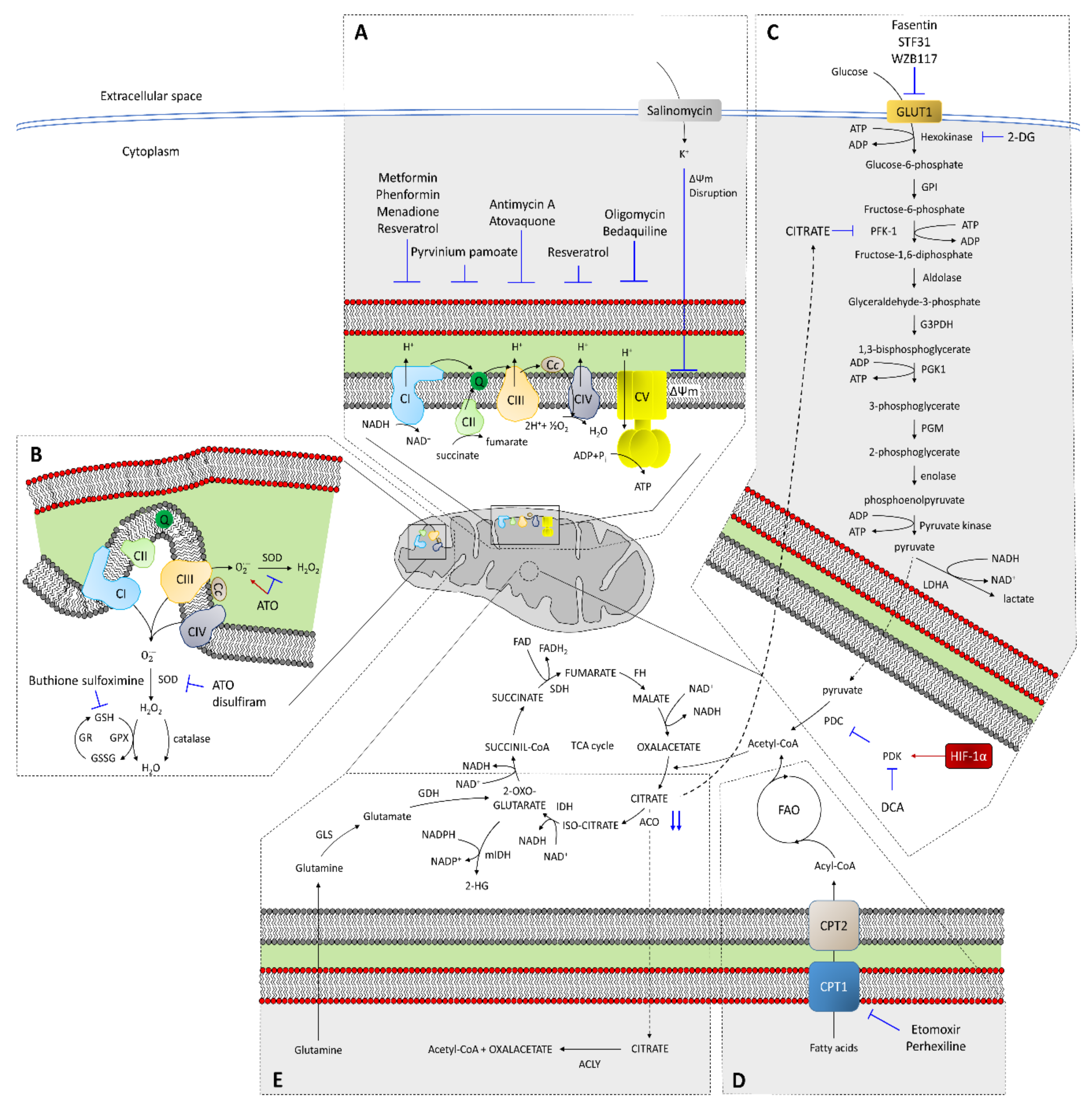
4.1. Role of the Mitochondrial Respiratory Chain in CSC Resistance
4.2. RNOS in CSC Resistance
4.3. Glycolysis
4.4. Fatty Acid Oxidation in CSC Resistance
4.5. Other Mitochondrial Metabolism Scenarios Implied in CSC Resistance
5. Horizontal Transference Connected to CSC Resistance
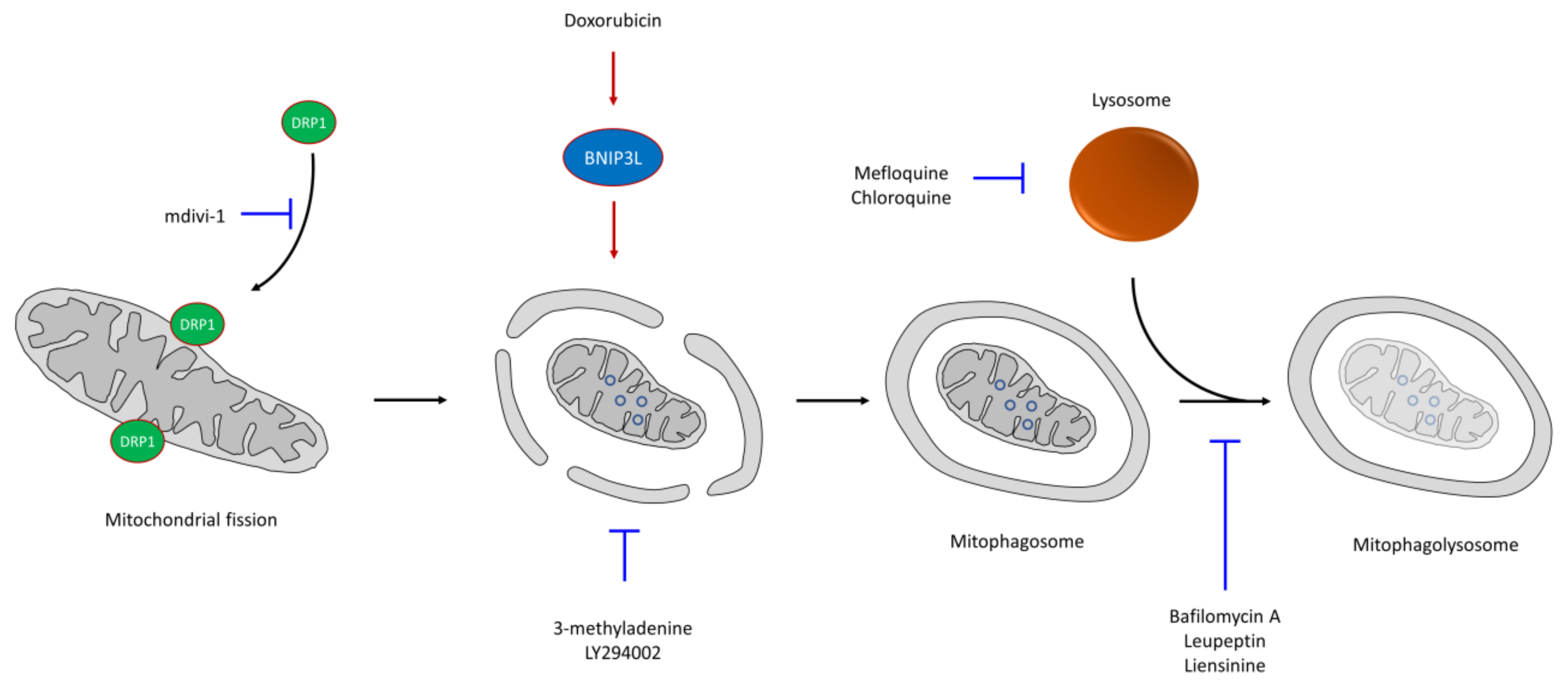
6. CSC Resistance in Mitophagy
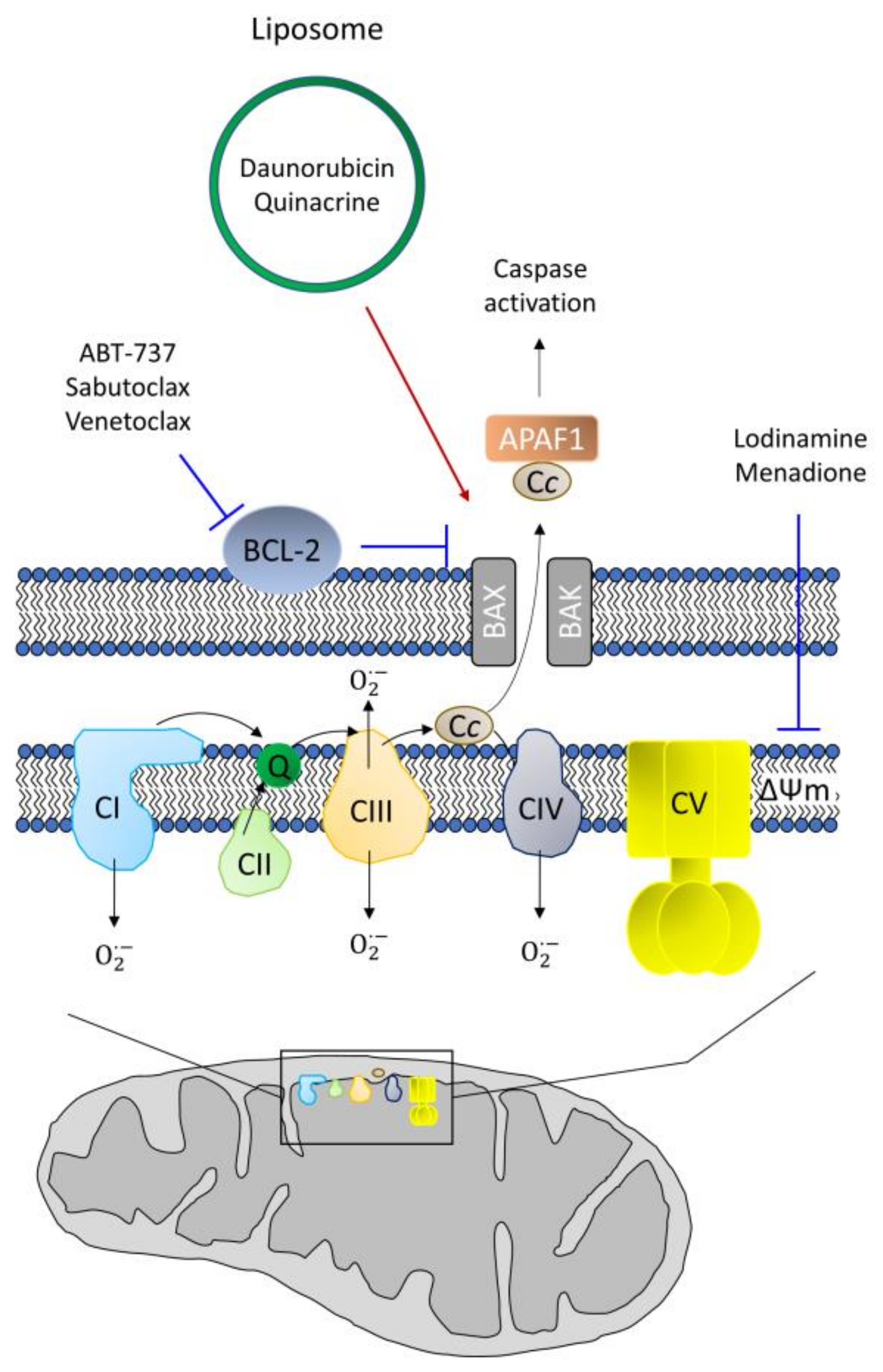
7. CSC Resistance in Apoptosis
8. Conclusions
Funding
Conflicts of Interest
References
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. In Stromal Immunology; Owens, B.M.J., Lakins, M.A., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 99–114. [Google Scholar]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, C.; Zhou, B.P. Metabolic reprogram associated with epithelial-mesenchymal transition in tumor progression and metastasis. Genes Dis. 2020, 7, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Pajonk, F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin. Cancer Biol. 2015, 31, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Dick, J.E. Cancer Stem Cells in Solid Tumors: An Overview. Semin. Radiat. Oncol. 2009, 19, 71–77. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Fu, L. Targeting cancer stem cells: A new therapy to cure cancer patients. Am. J. Cancer Res. 2012, 2, 340–356. [Google Scholar] [PubMed]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyakhovich, A.; Lleonart, M.E. Bypassing Mechanisms of Mitochondria-Mediated Cancer Stem Cells Resistance to Chemo- and Radiotherapy. Oxidative Med. Cell. Longev. 2016, 2016, 1716341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Studer, L.; Vera, E.; Cornacchia, D. Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell Stem Cell 2015, 16, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α; Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [Green Version]
- Paldino, E.; Tesori, V.; Casalbore, P.; Gasbarrini, A.; Puglisi, M.A. Tumor Initiating Cells and Chemoresistance: Which Is the Best Strategy to Target Colon Cancer Stem Cells? Biomed. Res. Int. 2014, 2014, 859871. [Google Scholar] [CrossRef]
- Chen, D.; Wang, C.-Y. Targeting cancer stem cells in squamous cell carcinoma. Precis. Clin. Med. 2019, 2, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-H.; Zhang, Q.-P. Leukemia stem cells in drug resistance and metastasis. Chin. Med. J. 2010, 123, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Frank, M.H. Cancer stem cells and human malignant melanoma. Pigment. Cell Melanoma Res. 2008, 21, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakariassen, P.Ø.; Immervoll, H.; Chekenya, M. Cancer stem cells as mediators of treatment resistance in brain tumors: Status and controversies. Neoplasia 2007, 9, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Rathod, M.; De, A. Targeting stem cells in the realm of drug-resistant breast cancer. Breast Cancer (Dove Med. Press) 2019, 11, 115–135. [Google Scholar] [CrossRef] [Green Version]
- Ercan, G.; Karlitepe, A.; Ozpolat, B. Pancreatic Cancer Stem Cells and Therapeutic Approaches. Anticancer Res. 2017, 37, 2761–2775. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal Adhesion Kinase and Wnt Signaling Regulate Human Ductal Carcinoma In Situ Stem Cell Activity and Response to Radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Coyle, K.M.; Vidovic, D.; Thomas, M.L.; Gujar, S.; Marcato, P. Hide-and-seek: The interplay between cancer stem cells and the immune system. Carcinogenesis 2016, 38, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Li, T.-S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagust, P.; de Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.L.; Francescangeli, F.; La Torre, F.; Zeuner, A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef] [Green Version]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Carcinogenesis, cancer therapy and chemoprevention. Cell Death Differ. 2005, 12, 592–602. [Google Scholar] [CrossRef] [Green Version]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug Transporters in Drug Efficacy and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef]
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. Biomed. Res. Int. 2015, 2015, 635745. [Google Scholar] [CrossRef] [Green Version]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Haigang, C.; Anna, J.Z.; Mingwei, C.; Johnson, J.L. ABC Transporter Inhibitors in Reversing Multidrug Resistance to Chemotherapy. Curr. Drug Targets 2015, 16, 1356–1371. [Google Scholar] [CrossRef]
- Pisco, A.O.; Jackson, D.A.; Huang, S. Reduced Intracellular Drug Accumulation in Drug-Resistant Leukemia Cells is Not Only Solely Due to MDR-Mediated Efflux but also to Decreased Uptake. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Y.; Hill, R.A.; Li, Y.-T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res. 2013, 117, 59–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klohs, W.D.; Steinkampf, R.W. The effect of lysosomotropic agents and secretory inhibitors on anthracycline retention and activity in multiple drug-resistant cells. Mol. Pharmacol. 1988, 34, 180–185. [Google Scholar]
- Mertins, S.D. Cancer stem cells: A systems biology view of their role in prognosis and therapy. Anticancer Drugs 2014, 25, 353–367. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.K.; Lin, Z.; Balliet, R.M.; Howell, A.; et al. Understanding the “lethal” drivers of tumor-stroma co-evolution. Cancer Biol. Ther. 2010, 10, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Et Biophys. Acta (Bba) Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Cannino, G.; Ciscato, F.; Masgras, I.; Sánchez-Martín, C.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333. [Google Scholar] [CrossRef]
- Obre, E.; Rossignol, R. Emerging concepts in bioenergetics and cancer research: Metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 2015, 59, 167–181. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Alptekin, A.; Ye, B.; Ding, H.-F. Transcriptional Regulation of Stem Cell and Cancer Stem Cell Metabolism. Curr. Stem Cell Rep. 2017, 3, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; Srinivasan, S.; Ruthel, G.; Kashina, A.K.; Carstens, R.P.; Mendoza, A.; Khanna, C.; Van Winkle, T.; Avadhani, N.G. Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Oncogene 2014, 33, 5238–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.-K.; Cheong, J.-H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharmacal Res. 2019, 42, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Jazwinski, S.M.; Kriete, A. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front. Physiol. 2012, 3, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Mejia, I.C.; Fajas, L. Cell cycle regulation of mitochondrial function. Curr. Opin. Cell Biol. 2015, 33, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Resistance to Cell Death and Its Modulation in Cancer Stem Cells. Crit. Rev. Oncog. 2016, 21, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, K.Z.Q.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Wu, S.-B.; Hong, C.-H.; Liao, W.-T.; Wu, C.-Y.; Chen, G.-S.; Wei, Y.-H.; Yu, H.-S. Aberrant cell proliferation by enhanced mitochondrial biogenesis via mtTFA in arsenical skin cancers. Am. J. Pathol. 2011, 178, 2066–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum. Mol. Genet. 2011, 20, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Moraes, C.T. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol. Oncol. 2011, 5, 399–409. [Google Scholar] [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting hypoxic cancer stem cells (CSCs) with Doxycycline: Implications for optimizing anti-angiogenic therapy. Oncotarget 2017, 8, 56126–56142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, L.; Zhang, F.; Vlashi, E. Doxycycline inhibits the cancer stem cell phenotype and epithelial-to-mesenchymal transition in breast cancer. Cell Cycle (Georget. Tex.) 2017, 16, 737–745. [Google Scholar] [CrossRef]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an Inhibitor of Mitochondrial Biogenesis, Effectively Reduces Cancer Stem Cells (CSCs) in Early Breast Cancer Patients: A Clinical Pilot Study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef]
- Chu, D.J.; Yao, D.E.; Zhuang, Y.F.; Hong, Y.; Zhu, X.C.; Fang, Z.R.; Yu, J.; Yu, Z.Y. Azithromycin enhances the favorable results of paclitaxel and cisplatin in patients with advanced non-small cell lung cancer. Genet. Mol. Res. 2014, 13, 2796–2805. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Bonuccelli, G.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 2017, 8, 67269–67286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, M.J.; Nguyen, D.; Kunkel, T.A.; Copeland, W.C. The Fidelity of Human DNA Polymerase γ with and without Exonucleolytic Proofreading and the p55 Accessory Subunit. J. Biol. Chem. 2001, 276, 38555–38562. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Heredia, J.M.; Carnero, A. Decoding Warburg’s hypothesis: Tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 2015, 6, 41582–41599. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, S.; Miyato, Y.; Shidara, Y.; Asoh, S.; Tokunaga, A.; Tajiri, T.; Ohta, S. Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 2009, 100, 1680–1687. [Google Scholar] [CrossRef]
- Tseng, L.-M.; Yin, P.-H.; Chi, C.-W.; Hsu, C.-Y.; Wu, C.-W.; Lee, L.-M.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. GenesChromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.C.; Higuchi, M. The awakening of an advanced malignant cancer: An insult to the mitochondrial genome. Biochim. Et Biophys. Acta (Bba) Gen. Subj. 2012, 1820, 652–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Naito, A.; Asano, T.; Evans, T.T.; Reddy, S.A.G.; Higuchi, M. Constitutive activation of AKT pathway inhibits TNF-induced apoptosis in mitochondrial DNA-deficient human myelogenous leukemia ML-1a. Cancer Lett. 2008, 268, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Chang, I.; Kim, J.-Y.; Kang, S.W.; Park, S.-H.; Singh, K.; Lee, M.-S. Resistance of Mitochondrial DNA-depleted Cells against Cell Death: Role of mitochondrial superoxide dismutase. J. Biol. Chem. 2004, 279, 7512–7520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, A.; Cook, C.C.; Mizumachi, T.; Wang, M.; Xie, C.-H.; Evans, T.T.; Kelly, T.; Higuchi, M. Progressive tumor features accompany epithelial-mesenchymal transition induced in mitochondrial DNA-depleted cells. Cancer Sci. 2008, 99, 1584–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, L.; Arbini, A.A.; Marra, E.; Greco, M. Mitochondrial DNA depletion reduces PARP-1 levels and promotes progression of the neoplastic phenotype in prostate carcinoma. Cell Oncol. 2008, 30, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Biswas, G.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mechanism of mitochondrial stress-induced resistance to apoptosis in mitochondrial DNA-depleted C2C12 myocytes. Cell Death Differ. 2005, 12, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Felipe-Abrio, B.; Carnero, A. The Tumor Suppressor Roles of MYBBP1A, a Major Contributor to Metabolism Plasticity and Stemness. Cancers 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Felipe-Abrio, B.; Verdugo-Sivianes, E.M.; Carnero, A. c-MYB- and PGC1a-dependent metabolic switch induced by MYBBP1A loss in renal cancer. Mol. Oncol. 2019, 13, 1519–1533. [Google Scholar] [CrossRef] [Green Version]
- Felipe-Abrio, B.; Verdugo-Sivianes, E.M.; Saez, C.; Carnero, A. Loss of MYBBP1A Induces Cancer Stem Cell Activity in Renal Cancer. Cancers 2019, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 915–992. [Google Scholar] [CrossRef] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-H.; Hung, P.-H.; Chen, Y.-J. CD44 is associated with the aggressive phenotype of nasopharyngeal carcinoma through redox regulation. Int. J. Mol. Sci. 2013, 14, 13266–13281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardman, P. Chemical Radiosensitizers for Use in Radiotherapy. Clin. Oncol. 2007, 19, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cipolleschi, M.G.; Marzi, I.; Santini, R.; Fredducci, D.; Vinci, M.C.; D’Amico, M.; Rovida, E.; Stivarou, T.; Torre, E.; Dello Sbarba, P.; et al. Hypoxia-resistant profile implies vulnerability of cancer stem cells to physiological agents, which suggests new therapeutic targets. Cell Cycle (Georget. Tex.) 2014, 13, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, A.; Lehninger, A.L. Bypasses of the antimycin A block of mitochondrial electron transport in relation to ubisemiquinone function. Biochim. Et Biophys. Acta (Bba) Bioenerg. 1984, 767, 120–129. [Google Scholar] [CrossRef]
- Lohberger, B.; Rinner, B.; Stuendl, N.; Absenger, M.; Liegl-Atzwanger, B.; Walzer, S.M.; Windhager, R.; Leithner, A. Aldehyde dehydrogenase 1, a potential marker for cancer stem cells in human sarcoma. PLoS ONE 2012, 7, e43664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Lo, M.; Moody, R.R.; Stevers, N.O.; Tinsley, S.L.; Sun, D. Doxycycline targets aldehyde dehydrogenase-positive breast cancer stem cells. Oncol. Rep. 2018, 39, 3041–3047. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Sun, L.; Mochly-Rosen, D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc. Res. 2010, 88, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Gasparre, G.; Kurelac, I.; Capristo, M.; Iommarini, L.; Ghelli, A.; Ceccarelli, C.; Nicoletti, G.; Nanni, P.; De Giovanni, C.; Scotlandi, K.; et al. A Mutation Threshold Distinguishes the Antitumorigenic Effects of the Mitochondrial Gene MTND1, an Oncojanus Function. Cancer Res. 2011, 71, 6220–6229. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci. Rep. 2019, 9, 5864. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.H.; Li, S.; Lu, G.; Xue, H.; Kim, D.H.; Zhu, J.-J.; Liu, Y. Metformin treatment reduces temozolomide resistance of glioblastoma cells. Oncotarget 2016, 7, 78787–78803. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.-H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Wa Cheng, K.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual Inhibition of Tumor Energy Pathway by 2-Deoxyglucose and Metformin Is Effective against a Broad Spectrum of Preclinical Cancer Models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García Rubiño, M.E.; Carrillo, E.; Ruiz Alcalá, G.; Domínguez-Martín, A.; Marchal, J.; Boulaiz, H. Phenformin as an Anticancer Agent: Challenges and Prospects. Int. J. Mol. Sci. 2019, 20, 3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Shim, J.-K.; Kang, J.H.; Choi, J.; Chang, J.H.; Kim, S.-Y.; Kang, S.-G. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro. Oncol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-T.; Su, C.-L.; Huang, C.-Y.F.; Lin, J.K.-Y.; Lee, W.-H.; Chang, P.M.H.; Kuo, Y.-L.; Liu, Y.-W.; Wang, L.-S.; Wu, C.-H.; et al. A preclinical evaluation of antimycin a as a potential antilung cancer stem cell agent. Evid Based Complement. Altern. Med. 2013, 2013, 910451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, C.R.; Tilkens, S.B.; Guan, H.; Garner, J.A.; Or, P.M.Y.; Chan, A.M. Differential sensitivities of glioblastoma cell lines towards metabolic and signaling pathway inhibitions. Cancer Lett. 2013, 336, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Chao, T.-K.; Chang, C.-C.; Yo, Y.-T.; Yu, M.-H.; Lai, H.-C. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS ONE 2013, 8, e74538. [Google Scholar] [CrossRef] [Green Version]
- Yo, Y.-T.; Lin, Y.-W.; Wang, Y.-C.; Balch, C.; Huang, R.-L.; Chan, M.W.Y.; Sytwu, H.-K.; Chen, C.-K.; Chang, C.-C.; Nephew, K.P.; et al. Growth Inhibition of Ovarian Tumor–Initiating Cells by Niclosamide. Mol. Cancer Ther. 2012, 11, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Diacon, A.H.; Pym, A.; Grobusch, M.P.; de los Rios, J.M.; Gotuzzo, E.; Vasilyeva, I.; Leimane, V.; Andries, K.; Bakare, N.; De Marez, T.; et al. Multidrug-Resistant Tuberculosis and Culture Conversion with Bedaquiline. N. Engl. J. Med. 2014, 371, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany N. Y.) 2016, 8, 1593–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes Costa, A.; Le Bachelier, C.; Mathieu, L.; Rotig, A.; Boneh, A.; De Lonlay, P.; Tarnopolsky, M.A.; Thorburn, D.R.; Bastin, J.; Djouadi, F. Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum. Mol. Genet. 2013, 23, 2106–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Zong, L.; Chen, X.; Jiang, Z.; Nan, L.; Li, J.; Duan, W.; Lei, J.; Zhang, L.; Ma, J.; et al. Resveratrol in the treatment of pancreatic cancer. Ann. N. Y. Acad. Sci. 2015, 1348, 10–19. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Naujokat, C.; Steinhart, R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol. 2012, 2012, 950658. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Jain, M.V.; Hallbeck, A.-L.; Roberg, K.; Lotfi, K.; Łos, M.J. Glucose starvation-mediated inhibition of salinomycin induced autophagy amplifies cancer cell specific cell death. Oncotarget 2015, 6, 10134–10145. [Google Scholar] [CrossRef]
- Jiang, J.; Li, H.; Qaed, E.; Zhang, J.; Song, Y.; Wu, R.; Bu, X.; Wang, Q.; Tang, Z. Salinomycin, as an autophagy modulator-- a new avenue to anticancer: A review. J. Exp. Clin. Cancer Res. 2018, 37, 26. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.-Q.; Zhen, Y.-F.; Zhang, Y.; Guo, Z.-X.; Dai, J.; Wang, X.-D. Salinomycin Activates AMP-Activated Protein Kinase-Dependent Autophagy in Cultured Osteoblastoma Cells: A Negative Regulator against Cell Apoptosis. PLoS ONE 2013, 8, e84175. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Ghavami, S.; Grabarek, J.; Kratz, G.; Wiechec, E.; Fredriksson, B.-A.; Rao Pariti, R.K.; Cieślar-Pobuda, A.; Panigrahi, S.; Łos, M.J. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: Differences between primary and cancer cells. Biochim. Et Biophys. Acta (Bba) Mol. Cell Res. 2013, 1833, 2057–2069. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fryknäs, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Påhlman, S.; Schughart, L.A.K.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; de Milito, A.; Olofsson, M.H.; Gullbo, J.; D’Arcy, P.; Linder, S. Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors. Int. J. Mol. Sci. 2015, 16, 27313–27326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive Oxygen Species as Intracellular Messengers During Cell Growth and Differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.C.; Yau, C. Ageing, oxidative stress and cancer: Paradigms in parallax. Nat. Rev. Cancer 2008, 8, 875–879. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.-G.; Lee, S.-H.; Kwak, M.-K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid. Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yang, J.; Li, L.; Sun, L.; Yi, X.; Han, X.; Si, W.; Yan, R.; Chen, Z.; Xie, G.; et al. PAAT, a novel ATPase and trans-regulator of mitochondrial ABC transporters, is critically involved in the maintenance of mitochondrial homeostasis. Faseb J. 2014, 28, 4821–4834. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Rolando, B.; Kopecka, J.; Campia, I.; Chegaev, K.; Lazzarato, L.; Federico, A.; Fruttero, R.; Ghigo, D. Mitochondrial-Targeting Nitrooxy-doxorubicin: A New Approach to Overcome Drug Resistance. Mol. Pharm. 2013, 10, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-J.; Tang, Q.; Li, Y.; Hu, B.-R.; Ming, Z.-Y.; Fu, Q.; Qian, J.-Q.; Xiang, J.-Z. Role of oxidative stress in the apoptosis of hepatocellular carcinoma induced by combination of arsenic trioxide and ascorbic acid. Acta Pharmacol. Sin. 2006, 27, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Aparicio, A.; Strasberg-Rieber, M.; Rieber, M. Disulfiram anti-cancer efficacy without copper overload is enhanced by extracellular H2O2 generation: Antagonism by tetrathiomolybdate. Oncotarget 2015, 6, 29771–29781. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Kumar, I.S.; Brown, S.; Kannappan, V.; Tawari, P.E.; Tang, J.Z.; Jiang, W.; Armesilla, A.L.; Darling, J.L.; Wang, W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br. J. Cancer 2013, 109, 1876–1885. [Google Scholar] [CrossRef] [Green Version]
- Sarsour, E.H.; Venkataraman, S.; Kalen, A.L.; Oberley, L.W.; Goswami, P.C. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell 2008, 7, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kirk, J.S.; Venkataraman, S.; Domann, F.E.; Zhang, H.J.; Schafer, F.Q.; Flanagan, S.W.; Weydert, C.J.; Spitz, D.R.; Buettner, G.R.; et al. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1alpha and vascular endothelial growth factor. Oncogene 2005, 24, 8154–8166. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High Thioredoxin Expression Is Associated with Resistance to Docetaxel in Primary Breast Cancer. Clin. Cancer Res. 2005, 11, 8425–8430. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Hosoi, F.; Yamaguchi-Iwai, Y.; Nakamura, H.; Masutani, H.; Ueda, S.; Nishiyama, A.; Takeda, S.; Wada, H.; Spyrou, G.; et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef] [Green Version]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat Res. 2016, 186, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic. Biol. Med. 2015, 88, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Harder, B.G.; Wong, P.K.; Lang, J.E.; Zhang, D.D. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol. Carcinog. 2015, 54, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, A.-M.; Ke, Z.-P.; Wang, J.-N.; Yang, J.-Y.; Chen, S.-Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Wang, J.; Sim, M.-S.; Liu, B.; Giuliano, A.; Barsoum, J.; Cui, X. Elesclomol, counteracted by Akt survival signaling, enhances the apoptotic effect of chemotherapy drugs in breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 311–321. [Google Scholar] [CrossRef]
- Leskovar, A.; Wegele, H.; Werbeck, N.D.; Buchner, J.; Reinstein, J. The ATPase Cycle of the Mitochondrial Hsp90 Analog Trap1. J. Biol. Chem. 2008, 283, 11677–11688. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of Tumor Cell Mitochondrial Homeostasis by an Organelle-Specific Hsp90 Chaperone Network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Song, H.Y.; Meli, M.; Colombo, G.; Beebe, K.; Scroggins, B.; Neckers, L.; Altieri, D.C. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Investig. 2009, 119, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Fan, T.W.M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [Green Version]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy Metabolism Characterization of a Novel Cancer Stem Cell-Like Line 3AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Dauer, P.; Gupta, V.; McGinn, O.; Arora, N.; Majumdar, K.; Uhlrich, C., 3rd; Dalluge, J.; Dudeja, V.; Saluja, A.; et al. Microenvironment mediated alterations to metabolic pathways confer increased chemo-resistance in CD133+ tumor initiating cells. Oncotarget 2016, 7, 56324–56337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Heredia, J.M.; Felipe-Abrio, B.; Cano, D.A.; Carnero, A. Genetic modification of hypoxia signaling in animal models and its effect on cancer. Clin. Transl. Oncol. 2015, 17, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.S.; Pereira, S.L.; Correia, M.; Gomes, A.; Perestrelo, T.; Ramalho-Santos, J. Differentiate or Die: 3-Bromopyruvate and Pluripotency in Mouse Embryonic Stem Cells. PLoS ONE 2015, 10, e0135617. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.-L.; Mackey, J.R.; Fulton, D.; et al. Metabolic Modulation of Glioblastoma with Dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Papandreou, I.; Sutphin, P.D.; Denko, N.C. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2007, 104, 9445–9450. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, R.; Yu, Z.; Li, R.; Li, J.; Zhao, X.; Song, S.; Liu, J.; Huang, G. Dichloroacetate restores drug sensitivity in paclitaxel-resistant cells by inducing citric acid accumulation. Mol. Cancer 2015, 14, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Et Biophys. Acta (Bba) Rev. Cancer 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Saccà, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.-M.; Lu, G.-D. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxidative Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.A.; Angka, L.; Rota, S.-G.; Hanlon, T.; Hurren, R.; Wang, X.M.; Gronda, M.; Bernard, D.; Minden, M.D.; Mitchell, A.; et al. Inhibition of Fatty Acid Oxidation with Avocatin B Selectively Targets AML Cells and Leukemia Stem Cells. Blood 2014, 124, 268. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44+CD24-/low breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-Z.; Ma, Y.; Zhou, Y.; Xu, L.-M.; Chen, X.-J.; Ding, W.-B.; Zou, H.-B. Autophagy contributes to the enrichment and survival of colorectal cancer stem cells under oxaliplatin treatment. Cancer Lett. 2015, 361, 128–136. [Google Scholar] [CrossRef]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.G.; Yang, J.C.; Kung, H.J.; Shi, X.B.; Tilki, D.; Lara, P.N., Jr.; DeVere White, R.W.; Gao, A.C.; Evans, C.P. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Pascolo, S. Time to use a dose of Chloroquine as an adjuvant to anti-cancer chemotherapies. Eur. J. Pharmacol. 2016, 771, 139–144. [Google Scholar] [CrossRef]
- Barnard, R.A.; Wittenburg, L.A.; Amaravadi, R.K.; Gustafson, D.L.; Thorburn, A.; Thamm, D.H. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014, 10, 1415–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xi, C.; Wang, W.; Yang, Y.; Qiu, Y.; Huang, Z. Autophagy plays an important role in triptolide-induced apoptosis in cardiomyocytes. Toxicol. Lett. 2015, 236, 168–183. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Luo, L.; Guo, C.-Y.; Goto, S.; Urata, Y.; Shao, J.-H.; Li, T.-S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef]
- Marino, M.L.; Fais, S.; Djavaheri-Mergny, M.; Villa, A.; Meschini, S.; Lozupone, F.; Venturi, G.; Della Mina, P.; Pattingre, S.; Rivoltini, L.; et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010, 1, e87. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Hill, D.K.; Andrejeva, G.; Boult, J.K.R.; Troy, H.; Fong, A.C.L.F.W.T.; Orton, M.R.; Panek, R.; Parkes, H.G.; Jafar, M.; et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br. J. Cancer 2014, 111, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornet-Masana, J.M.; Banús-Mulet, A.; Carbó, J.M.; Torrente, M.Á.; Guijarro, F.; Cuesta-Casanovas, L.; Esteve, J.; Risueño, R.M. Dual lysosomal-mitochondrial targeting by antihistamines to eradicate leukaemic cells. EBioMedicine 2019, 47, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.-M.; Hu, X.; Ming, Q.-L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radogna, F.; Cerella, C.; Gaigneaux, A.; Christov, C.; Dicato, M.; Diederich, M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 2016, 35, 3839–3853. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Rosdah, A.A.; Holien, J.K.; Delbridge, L.M.D.; Dusting, G.J.; Lim, S.Y. Mitochondrial fission - a drug target for cytoprotection or cytodestruction? Pharm. Res. Perspect 2016, 4, e00235. [Google Scholar] [CrossRef]
- Kong, B.; Tsuyoshi, H.; Orisaka, M.; Shieh, D.-B.; Yoshida, Y.; Tsang, B.K. Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann. N. Y. Acad. Sci. 2015, 1350, 1–16. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254–13275. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Wang, J.; Roginskaya, V.; McDermott, L.A.; Edwards, R.P.; Stolz, D.B.; Llambi, F.; Green, D.R.; Van Houten, B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180–4194. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondrial control of apoptosis: An introduction. Biochem. Biophys. Res. Commun. 2003, 304, 433–435. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yao, H.-J.; Yu, Y.; Zhang, Y.; Li, R.-J.; Ju, R.-J.; Wang, X.-X.; Sun, M.-G.; Shi, J.-F.; Lu, W.-L. Mitochondrial targeting liposomes incorporating daunorubicin and quinacrine for treatment of relapsed breast cancer arising from cancer stem cells. Biomaterials 2012, 33, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- De Fabritiis, P.; Sandrelli, A.; Covelli, A.; Simone, F.; De Felice, L.; Pulsoni, A.; Zupi, G.; Mandelli, F. In vitro pharmacological purging of human bone marrow is enhanced by the use of lonidamine. Exp. Mol. Pathol. 1989, 50, 210–219. [Google Scholar] [CrossRef]
- Solary, E.; Bettaieb, A.; Dubrez-Daloz, L.; Corcos, L. Mitochondria as a Target for Inducing Death of Malignant Hematopoietic Cells. Leuk. Lymphoma 2003, 44, 563–574. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-H.; Scadden, D.T. Harnessing the apoptotic programs in cancer stem-like cells. EMBO Rep. 2015, 16, 1084–1098. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yagüe, E.; Zhao, J.; Wang, L.; Bai, J.; Yang, Q.; Pan, T.; Zhao, H.; Liu, J.; Zhang, J. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. 2018, 423, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Bhola, P.D.; Letai, A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci. Transl. Med. 2019, 11, eaax2863. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, C.; Raspaglio, G.; Mozzetti, S.; Distefano, M.; Filippetti, F.; Martinelli, E.; Ferrandina, G.; Gallo, D.; Ranelletti, F.O.; Scambia, G. Bcl-2 Down-Regulation Is a Novel Mechanism of Paclitaxel Resistance. Mol. Pharmacol. 2003, 64, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Hamo, R.; Zilberberg, A.; Cohen, H.; Bahar-Shany, K.; Wachtel, C.; Korach, J.; Aviel-Ronen, S.; Barshack, I.; Barash, D.; Levanon, K.; et al. Resistance to paclitaxel is associated with a variant of the gene BCL2 in multiple tumor types. Npj Precis. Oncol. 2019, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Mimata, H.; Takeuchi, Y.; Yamamoto, H.; Miyamoto, E.; Nomura, Y. The X-linked inhibitor of apoptosis protein inhibits taxol-induced apoptosis in LNCaP cells. Urol. Res. 2003, 31, 37–44. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. https://doi.org/10.3390/cells9071693
García-Heredia JM, Carnero A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells. 2020; 9(7):1693. https://doi.org/10.3390/cells9071693
Chicago/Turabian StyleGarcía-Heredia, José Manuel, and Amancio Carnero. 2020. "Role of Mitochondria in Cancer Stem Cell Resistance" Cells 9, no. 7: 1693. https://doi.org/10.3390/cells9071693
APA StyleGarcía-Heredia, J. M., & Carnero, A. (2020). Role of Mitochondria in Cancer Stem Cell Resistance. Cells, 9(7), 1693. https://doi.org/10.3390/cells9071693