Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution



Abstract
:1. Introduction
2. Types of Inaccurate Double-Strand Break Repair
2.1. Classical Nonhomologous End-Joining (cNHEJ): A First Responder
2.2. Alternative End-Joining: A Quick and Dirty Fix
2.3. Alt-EJ Can be Defined by Repair Outcomes
2.4. Alt-EJ Can be Defined by Genetic Requirements
2.5. The Mechanism of TMEJ
2.6. When Homologous Recombination “Goes off the Rails”
2.7. Break-Induced Replication
2.8. Single-Strand Annealing: Not Just Extreme MMEJ
3. Factors That Affect Repair Pathway Choice
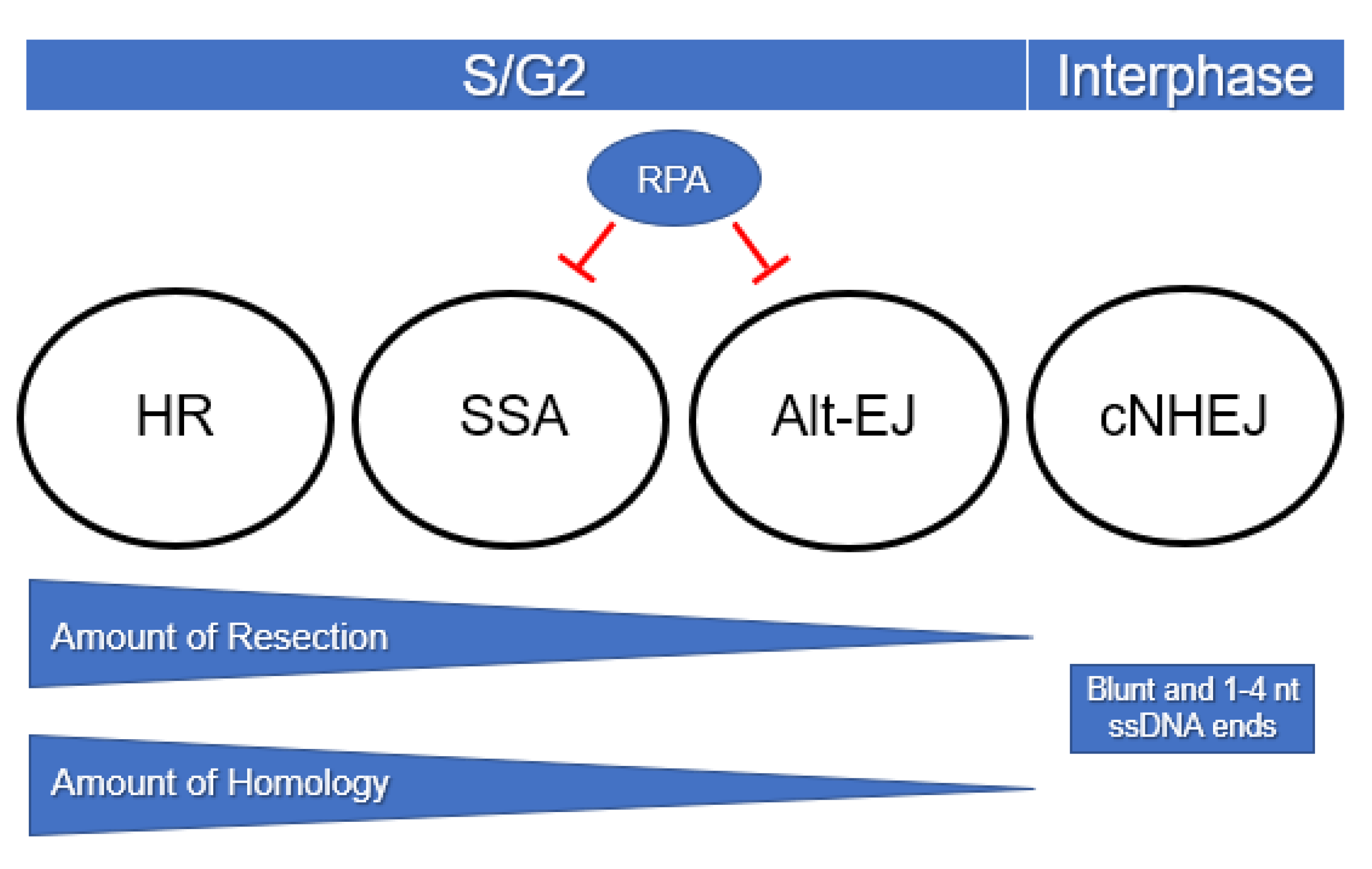
3.1. Cell Cycle Position
3.2. Resection
3.3. RPA
3.4. Microhomology
4. How Error-Prone Repair Relates to Genome Evolution
4.1. Alt-EJ Promotes Genome Evolution
4.2. Genome Compaction
4.3. Genome Expansion
4.4. Error-Prone Repair Contributes to Genome Evolution in Cancer
4.5. Genome Rearrangements
4.6. Chromothripsis
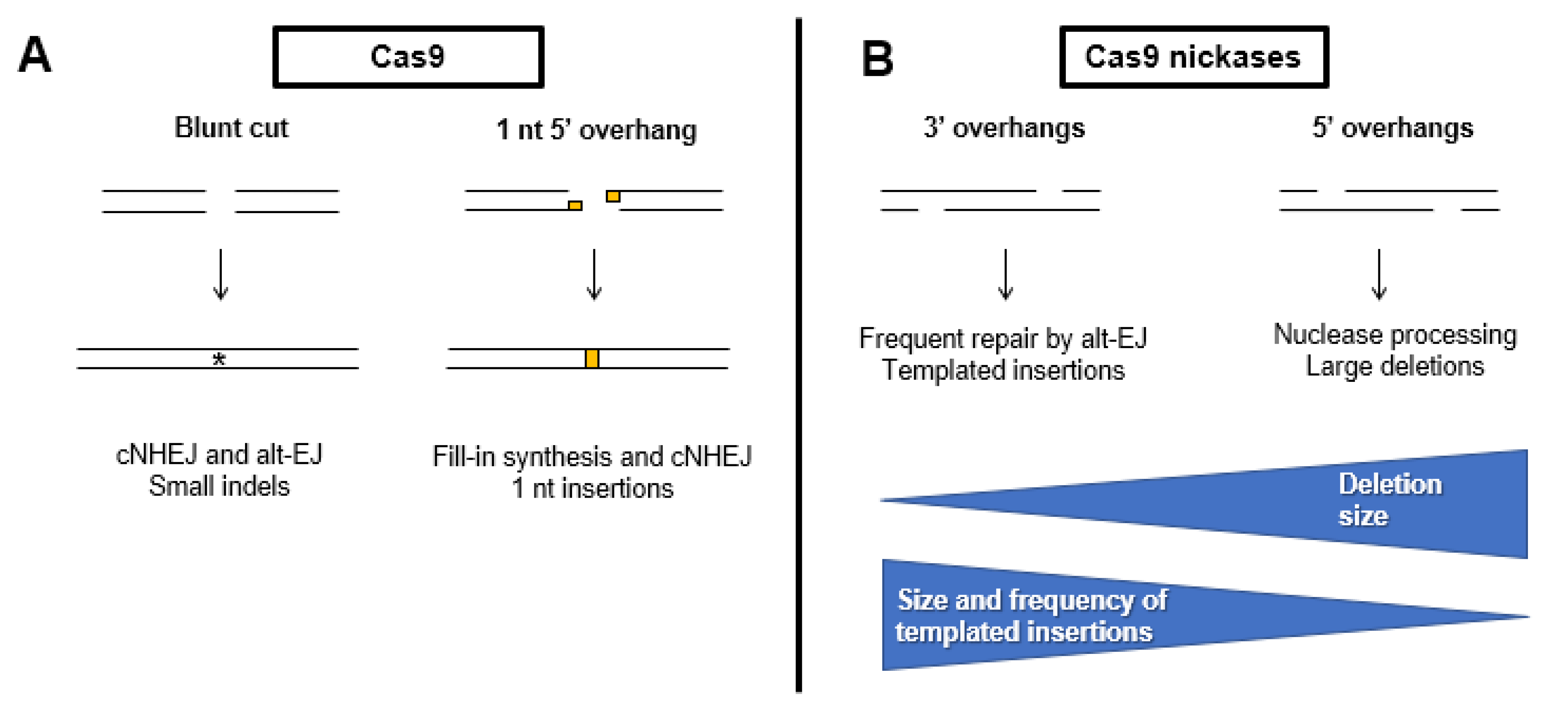
4.7. Repair Pathway Choice in the Context of Genome Editing
5. Conclusions and Remaining Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schendel, R.; Roerink, S.F.; Portegijs, V.; Heuvel, S.V.D.; Tijsterman, M. Polymerase Θ is a key driver of genome evolution and of CRISPR/Cas9-mediated mutagenesis. Nat. Commun. 2015, 6, 7394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Leeman, J.E.; Li, Y.; Bell, A.; Hussain, S.S.; Majumdar, R.; Rong-Mullins, X.; Blecua, P.; Damerla, R.; Narang, H.; Ravindran, P.T.; et al. Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc. Natl. Acad. Sci. USA 2019, 116, 21573–21579. [Google Scholar] [CrossRef] [Green Version]
- Higgins, G.S.; Harris, A.L.; Prevo, R.; Helleday, T.; McKenna, W.G.; Buffa, F.M. Overexpression of POLQ Confers a Poor Prognosis in Early Breast Cancer Patients. Oncotarget 2010, 1, 175–184. [Google Scholar] [CrossRef]
- Deriano, L.; Roth, D.B. Modernizing the Nonhomologous End-Joining Repertoire: Alternative and Classical NHEJ Share the Stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef]
- Krenning, L.; Berg, J.V.D.; Medema, R. Life or Death after a Break: What Determines the Choice? Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef]
- Wyatt, D.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R.; Gu, J.; Lu, H.; Shimazaki, N.; Tsai, A.G. Nonhomologous DNA End Joining (NHEJ) and Chromosomal Translocations in Humans. Subcell. Biochem. 2009, 50, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, D.; LeCain, M.; Sheer, D. The role of microhomology in genomic structural variation. Trends Genet. 2014, 30, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.-B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corneo, B.; Wendland, R.L.; Deriano, L.; Cui, X.; Klein, I.A.; Wong, S.-Y.; Arnal, S.; Holub, A.J.; Weller, G.R.; Pancake, B.A.; et al. Rag mutations reveal robust alternative end joining. Nature 2007, 449, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; He, J.; Razavian, N.; Berns, M.W.; Wu, X.; Hwang, P.Y.-H.; Wang, H. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Symington, L. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gómez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Roerink, S.F.; Van Schendel, R.; Tijsterman, M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res. 2014, 24, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, M.J.; Wood, R.D. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair 2013, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.-L.; Kim, E.M.; Haber, J.E.; Lee, S.E. Yeast Mre11 and Rad1 Proteins Define a Ku-Independent Mechanism to Repair Double-Strand Breaks Lacking Overlapping End Sequences. Mol. Cell. Biol. 2003, 23, 8820–8828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Lee, S.E. Saccharomyces cerevisiaeSae2- and Tel1-Dependent Single-Strand DNA Formation at DNA Break Promotes Microhomology-Mediated End Joining. Genetics 2007, 176, 2003–2014. [Google Scholar] [CrossRef] [Green Version]
- Pannunzio, N.R.; Li, S.; Watanabe, G.; Lieber, M.R. Non-homologous end joining often uses microhomology: Implications for alternative end joining. DNA Repair 2014, 17, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.M.; McVey, M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010, 38, 5706–5717. [Google Scholar] [CrossRef] [Green Version]
- Khodaverdian, V.Y.; Hanscom, T.; Yu, A.M.; Yu, T.L.; Mak, V.; Brown, A.J.; Roberts, S.A.; McVey, M. Secondary structure forming sequences drive SD-MMEJ repair of DNA double-strand breaks. Nucleic Acids Res. 2017, 45, 12848–12861. [Google Scholar] [CrossRef] [Green Version]
- Tsabar, M.; Haber, J.E. Chromatin modifications and chromatin remodeling during DNA repair in budding yeast. Curr. Opin. Genet. Dev. 2013, 23, 166–173. [Google Scholar] [CrossRef]
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar] [CrossRef] [Green Version]
- Paul, K.; Wang, M.; Mladenov, E.; Bencsik-Theilen, A.; Bednar, T.; Wu, W.; Arakawa, H.; Iliakis, G. DNA Ligases I and III Cooperate in Alternative Non-Homologous End-Joining in Vertebrates. PLoS ONE 2013, 8, e59505. [Google Scholar] [CrossRef]
- Chan, S.H.; Yu, A.M.; McVey, M. Dual Roles for DNA Polymerase Theta in Alternative End-Joining Repair of Double-Strand Breaks in Drosophila. PLoS Genet. 2010, 6, e1001005. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.E.; Averill, A.M.; Aller, P.; Wood, R.D.; Doublié, S. Human DNA polymerase theta grasps the primer terminus to mediate DNA repair. Nat. Struct. Mol. Biol. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, S.; Suzuki, T.; Ohto, M.-A.; Urawa, H.; Horiuchi, T.; Nakamura, K.; Morikami, A. Arabidopsis TEBICHI, with Helicase and DNA Polymerase Domains, Is Required for Regulated Cell Division and Differentiation in Meristems[W][OA]. Plant Cell 2006, 18, 879–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, J.; Kool, H.; Schendel, R.; Tijsterman, M. Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J. 2017, 36, 3634–3649. [Google Scholar] [CrossRef]
- Zelensky, A.N.; Schimmel, J.; Kool, H.; Kanaar, R.; Tijsterman, M. Inactivation of Pol theta and C-NHEJ eliminates off-target integration of exogenous DNA. Nat. Commun. 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Yang, W. Template and primer requirements for DNA Pol theta-mediated end joining. Proc. Natl. Acad. Sci. USA 2018, 115, 7747–7752. [Google Scholar] [CrossRef] [Green Version]
- Carvajal-Garcia, J.; Cho, J.-E.; Carvajal-Garcia, P.; Feng, W.; Wood, R.D.; Sekelsky, J.; Gupta, G.P.; Roberts, S.A.; Ramsden, D.A. Mechanistic basis for microhomology identification and genome scarring by polymerase theta. Proc. Natl. Acad. Sci. USA 2020, 117, 8476–8485. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, A.Y.; Rusanov, T.; Kent, T.; Siddique, L.A.; Pomerantz, R.T. Polymerase theta-helicase efficiently unwinds DNA and RNA-DNA hybrids. J. Biol. Chem. 2018, 293, 5259–5269. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Kent, T.; Deng, S.K.; McDevitt, S.; Kashkina, E.; Hoang, T.M.; Pomerantz, R.T.; Sfeir, A. The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 2017, 24, 1116–1123. [Google Scholar] [CrossRef]
- Beagan, K.; Armstrong, R.L.; Witsell, A.; Roy, U.; Renedo, N.; Baker, A.E.; Schärer, O.D.; McVey, M. Drosophila DNA polymerase theta utilizes both helicase-like and polymerase domains during microhomology-mediated end joining and interstrand crosslink repair. PLoS Genet. 2017, 13, e1006813. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yucel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. Polymerase Theta Inhibition Kills Homologous Recombination Deficient Tumors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, J.M.; Graves, T.; Newman, T.L.; Fulton, R.; Hayden, H.S.; Malig, M.; Kallicki, J.; Kaul, R.; Wilson, R.K.; Eichler, E.E. A Human Genome Structural Variation Sequencing Resource Reveals Insights into Mutational Mechanisms. Cell 2010, 143, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [CrossRef] [PubMed] [Green Version]
- Zepeda, C.; Lemus, T.; Yáñez, O.; García, D.; Valle-Garcia, D.; Meza-Sosa, K.F.; Gutiérrez-Arcelus, M.; Ortiz, Y.M.; Vidaña, R.D.; Gonzaga-Jauregui, C.; et al. Identical repeated backbone of the human genome. BMC Genom. 2010, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- White, T.B.; Morales, M.E.; Deininger, P.L. Alu elements and DNA double-strand break repair. Mob. Genet. Elem. 2015, 5, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Montagna, M.; Santacatterina, M.; Torri, A.; Menin, C.; Zullato, D.; Chieco-Bianchi, L.; D’Andrea, E. Identification of a 3 kb Alu-mediated BRCA1 gene rearrangement in two breast/ovarian cancer families. Oncogene 1999, 18, 4160–4165. [Google Scholar] [CrossRef] [Green Version]
- Kramara, J.; Osia, B.; Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef] [Green Version]
- Signon, L.; Malkova, A.; Naylor, M.L.; Klein, H.; Haber, J.E. Genetic Requirements for RAD51- andRAD54-Independent Break-Induced Replication Repair of a Chromosomal Double-Strand Break. Mol. Cell. Biol. 2001, 21, 2048–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantino, L.; Sotiriou, S.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.-P.; Halazonetis, T.D. Break-Induced Replication Repair of Damaged Forks Induces Genomic Duplications in Human Cells. Science 2013, 343, 88–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Beach, A.; Haber, J.E. Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol. Cell 2017, 65, 515–526.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.; Sterling, J.F.; Roberts, S.A.; Bhagwat, A.S.; Resnick, M.A.; Gordenin, D.A. Base Damage within Single-Strand DNA Underlies In Vivo Hypermutability Induced by a Ubiquitous Environmental Agent. PLoS Genet. 2012, 8, e1003149. [Google Scholar] [CrossRef]
- Zhang, F.; Khajavi, M.; Connolly, A.; Towne, C.F.; Batish, S.D.; Lupski, J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009, 41, 849–853. [Google Scholar] [CrossRef] [Green Version]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shim, E.Y.; Davis, M.; Lee, S.E. Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair 2009, 8, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Emerson, C.H.; Bertuch, A.A. Consider the workhorse: Nonhomologous end-joining in budding yeast. Biochem. Cell Biol. 2016, 94, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Stucki, M. How Cells Respond to DNA Breaks in Mitosis. Trends Biochem. Sci. 2020, 45, 321–331. [Google Scholar] [CrossRef]
- Giunta, S.; Belotserkovskaya, R.; Jackson, S.P. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010, 190, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, N.; Rajendra, E.; Su, X.; Jeyasekharan, A.D.; Mahen, R.; Venkitaraman, A.R. The Carboxyl Terminus of Brca2 Links the Disassembly of Rad51 Complexes to Mitotic Entry. Curr. Biol. 2009, 19, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Barral, P.; Vannier, J.-B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Du, Z.; Wang, Y.; Feng, Z.; Fan, P.; Yan, C.; Willers, H.; Zhang, J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015, 43, 1659–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, O.; Naumann, S.C.; Diemer-Biehs, R.; Künzel, J.; Steinlage, M.; Conrad, S.; Makharashvili, N.; Wang, J.; Feng, L.; Lopez, B.S.; et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014, 206, 877–894. [Google Scholar] [CrossRef]
- Reginato, G.; Cannavo, E.; Cejka, P. Physiological protein blocks direct the Mre11–Rad50–Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017, 31, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.-L.; Peters, T.J.; Khoury, A.; et al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun. 2019, 10, 416. [Google Scholar] [CrossRef]
- Eapen, V.V.; Sugawara, N.; Tsabar, M.; Wu, W.-H.; Haber, J.E. The Saccharomyces cerevisiae Chromatin Remodeler Fun30 Regulates DNA End Resection and Checkpoint Deactivation. Mol. Cell. Biol. 2012, 32, 4727–4740. [Google Scholar] [CrossRef] [Green Version]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.; Aramayo, R.J.; Pokhrel, N.; Caldwell, C.; Kaplan, J.A.; Perera, R.; Spies, M.; Antony, E.; Zhang, X. A structural and dynamic model for the assembly of Replication Protein A on single-stranded DNA. Nat. Commun. 2018, 9, 5447. [Google Scholar] [CrossRef]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Symington, L. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejka, P.; Cannavo, E.; Polaczek, P.; Masuda-Sasa, T.; Pokharel, S.; Campbell, J.L.; Kowalczykowski, S.C. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 2010, 467, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Gibb, B.; De Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, D.D.; Lee, K.; Deem, A.; Shim, E.Y.; Malkova, A.; Lee, S.E. Microhomology Directs Diverse DNA Break Repair Pathways and Chromosomal Translocations. PLoS Genet. 2012, 8, e1003026. [Google Scholar] [CrossRef] [Green Version]
- Decottignies, A. Microhomology-Mediated End Joining in Fission Yeast Is Repressed by Pku70 and Relies on Genes Involved in Homologous Recombination. Genetics 2007, 176, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Ji, J.-H.; Yoon, K.; Che, J.; Seol, J.-H.; Lee, S.E.; Shim, E.Y. Microhomology Selection for Microhomology Mediated End Joining in Saccharomyces cerevisiae. Genes 2019, 10, 284. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Beach, A.; Li, K.; Haber, J.E. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 2017, 544, 377–380. [Google Scholar] [CrossRef]
- Clejan, I.; Boerckel, J.; Ahmed, S. Developmental Modulation of Nonhomologous End Joining in Caenorhabditis elegans. Genetics 2006, 173, 1301–1317. [Google Scholar] [CrossRef] [Green Version]
- Joyce, E.F.; Paul, A.; Chen, K.E.; Tanneti, N.; McKim, K. Multiple Barriers to Nonhomologous DNA End Joining During Meiosis in Drosophila. Genetics 2012, 191, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manheim, E.; Jang, J.K.; Dominic, D.; McKim, K.S. Cytoplasmic Localization and Evolutionary Conservation of MEI-218, a Protein Required for Meiotic Crossing-over in Drosophila. Mol. Biol. Cell 2002, 13, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, G.; Cao, H.X.; Reiss, B.; Schubert, I. Deletion-bias in DNA double-strand break repair differentially contributes to plant genome shrinkage. New Phytol. 2017, 214, 1712–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Henriet, S.; Chourrout, D. Prevalence of Mutation-Prone Microhomology-Mediated End Joining in a Chordate Lacking the c-NHEJ DNA Repair Pathway. Curr. Biol. 2018, 28, 3337–3341.e4. [Google Scholar] [CrossRef]
- Glover, L.; Jun, J.; Horn, D. Microhomology-mediated deletion and gene conversion in African trypanosomes. Nucleic Acids Res. 2010, 39, 1372–1380. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Orgiler, A.; Martínez-Jiménez, M.I.; Alonso, A.; Alcolea, P.J.A.; Requena, J.M.; Thomas, M.C.; Blanco, L.; Larraga, V. A putative Leishmania DNA polymerase theta protects the parasite against oxidative damage. Nucleic Acids Res. 2016, 44, 4855–4870. [Google Scholar] [CrossRef] [Green Version]
- Hooks, K.B.; Delneri, D.; Griffiths-Jones, S. Intron Evolution in Saccharomycetaceae. Genome Biol. Evol. 2014, 6, 2543–2556. [Google Scholar] [CrossRef] [Green Version]
- Hu, K. Intron exclusion and the mystery of intron loss. FEBS Lett. 2006, 580, 6361–6365. [Google Scholar] [CrossRef] [Green Version]
- Yenerall, P.; Krupa, B.; Zhou, L. Mechanisms of intron gain and loss in Drosophila. BMC Evol. Biol. 2011, 11, 364. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Huang, Q.; Gao, D.; Wang, J.; Lang, Y.; Liu, T.; Li, B.; Bai, Z.; Goicoechea, J.L.; Liang, C.; et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nat. Commun. 2013, 4, 1595. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, A.; Jasin, M.; Chartrand, P. A Double-Strand Break in a Chromosomal LINE Element Can Be Repaired by Gene Conversion with Various Endogenous LINE Elements in Mouse Cells. Mol. Cell. Biol. 2000, 20, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Lee, J.; Meyer, T.J.; Remedios, P.; Goodwin, L.; Batzer, M.A. L1 recombination-associated deletions generate human genomic variation. Proc. Natl. Acad. Sci. USA 2008, 105, 19366–19371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, J.N.; Bennetzen, J.L. Natural insertions in rice commonly form tandem duplications indicative of patch-mediated double-strand break induction and repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6684–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, A.H.; Timmis, J. The Origin and Characterization of New Nuclear Genes Originating from a Cytoplasmic Organellar Genome. Mol. Biol. Evol. 2011, 28, 2019–2028. [Google Scholar] [CrossRef] [Green Version]
- Martin, W.F.; Rujan, T.; Richly, E.; Hansen, A.; Cornelsen, S.; Lins, T.; Leister, D.; Stoebe, B.; Hasegawa, M.; Penny, D. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. USA 2002, 99, 12246–12251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, C.; Ahmadinejad, N.; Wiegand, C.; Rotte, C.; Sebastiani, F.; Gelius-Dietrich, G.; Henze, K.; Kretschmann, E.; Richly, E.; Leister, D.; et al. A Genome Phylogeny for Mitochondria Among—Proteobacteria and a Predominantly Eubacterial Ancestry of Yeast Nuclear Genes. Mol. Biol. Evol. 2004, 21, 1643–1660. [Google Scholar] [CrossRef]
- Hazkani-Covo, E.; Covo, S. Numt-Mediated Double-Strand Break Repair Mitigates Deletions during Primate Genome Evolution. PLoS Genet. 2008, 4, e1000237. [Google Scholar] [CrossRef] [Green Version]
- Puertas, M.; Gonzalez-Sanchez, M. Insertions of mitochondrial DNA into the nucleus—Effects and role in cell evolution. Genome 2020. [Google Scholar] [CrossRef]
- Kellis, M.; Birren, B.W.; Lander, E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 2004, 428, 617–624. [Google Scholar] [CrossRef]
- Muramoto, N.; Oda, A.; Tanaka, H.; Nakamura, T.; Kugou, K.; Suda, K.; Kobayashi, A.; Yoneda, S.; Ikeuchi, A.; Sugimoto, H.; et al. Phenotypic diversification by enhanced genome restructuring after induction of multiple DNA double-strand breaks. Nat. Commun. 2018, 9, 1995. [Google Scholar] [CrossRef]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenner, A.; Sartori, A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Volkova, N.V.; Meier, B.; González-Huici, V.; Bertolini, S.; Gonzalez, S.; Vöhringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Zámborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Póti, Á.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017, 36, 5085–5086. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [Green Version]
- Lemée, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.-J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.-L.; LeDuc, C.; et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, J.; Serafim, R.; Ketchum, S.; Ferreira, C.G.; Liu, J.C.; Coe, K.A.; Price, B.D.; Yusufzai, T. Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its N terminus. Nucleic Acids Res. 2018, 46, 3891–3905. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, S.; Pellegrini, M.; Shuda, K.; Fernandez-Capetillo, O.; Liu, Y.; Martin, B.K.; Burkett, S.; Southon, E.; Pati, D.; Tessarollo, L.; et al. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J. Cell Biol. 2007, 176, 581–592. [Google Scholar] [CrossRef]
- Reh, W.A.; Nairn, R.S.; Lowery, M.P.; Vasquez, K.M. The homologous recombination protein RAD51D protects the genome from large deletions. Nucleic Acids Res. 2017, 45, 1835–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, L.A.; Robert, C.; Rapoport, A.P.; Gojo, I.; Baer, M.R.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene 2012, 32, 1784–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallmyr, A.; Tomkinson, A.E.; Rassool, F.V. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: Consequences for the repair of DNA double-strand breaks. Blood 2008, 112, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges. Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [Green Version]
- Bentley, J.; Diggle, C.P.; Harnden, P.; Knowles, M.A.; Kiltie, A.E. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004, 32, 5249–5259. [Google Scholar] [CrossRef] [Green Version]
- Difilippantonio, M.J.; Petersen, S.; Chen, H.T.; Johnson, R.; Jasin, M.; Kanaar, R.; Ried, T.; Nussenzweig, A. Evidence for Replicative Repair of DNA Double-Strand Breaks Leading to Oncogenic Translocation and Gene Amplification. J. Exp. Med. 2002, 196, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Mills, K.D.; Ferguson, D.O.; Lee, C.; Manis, J.; Fleming, J.; Gao, Y.; Morton, C.C.; Alt, F.W. Unrepaired DNA Breaks in p53-Deficient Cells Lead to Oncogenic Gene Amplification Subsequent to Translocations. Cell 2002, 109, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Gostissa, M.; Yan, C.T.; Goff, P.; Hickernell, T.; Hansen, E.; Difilippantonio, S.; Wesemann, D.R.; Zarrin, A.A.; Rajewsky, K.; et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature 2009, 460, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Tobin, L.A.; Robert, C.; Nagaria, P.; Chumsri, S.; Twaddell, W.; Ioffe, O.B.; Greco, G.E.; Brodie, A.H.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol. Cancer Res. 2011, 10, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, E.A.; Lu, F.; Bashllari, D.; Wang, L.; Opipari, A.W.; Opipari, V.P. Alternative NHEJ Pathway Components Are Therapeutic Targets in High-Risk Neuroblastoma. Mol. Cancer Res. 2015, 13, 470–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Hu, X.; Chen, L.; Cao, A.-Y.; Yu, K.-D.; Shi, T.-Y.; Kuang, X.-Y.; Shi, W.-B.; Ling, H.; Li, S.; et al. A recessive variant of XRCC4 predisposes to non- BRCA1/2 breast cancer in chinese women and impairs the DNA damage response via dysregulated nuclear localization. Oncotarget 2014, 5, 12218–12232. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Zhang, Y.; Lee, S.E. Saccharomyces cerevisiae ATM orthologue suppresses break-induced chromosome translocations. Nature 2008, 454, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Javadekar, S.M.; Raghavan, S.C. Snaps and mends: DNA breaks and chromosomal translocations. FEBS J. 2015, 282, 2627–2645. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.; Fu, B.X.; Heyer, W.D. DNA polymerases delta and lambda cooperate in repairing double-strand breaks by microhomology-mediated end-joining in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2015, 112, E6907–E6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, E.; Simsek, D.; Tomishima, M.; DeKelver, R.; Choi, V.M.; Gregory, P.; Urnov, F.; Weinstock, D.M.; Jasin, M. Chromosomal translocations induced at specified loci in human stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10620–10625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; McBride, D.J.; Lin, M.-L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.E.; Oh, S.; Grimstead, J.W.; Zimbric, J.; Roger, L.; Heppel, N.H.; Ashelford, K.E.; Liddiard, K.; Hendrickson, E.A.; Baird, D.M. Escape from Telomere-Driven Crisis Is DNA Ligase III Dependent. Cell Rep. 2014, 8, 1063–1076. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Kloosterman, W.P.; Tavakoli-Yaraki, M.; Van Roosmalen, M.J.; Van Binsbergen, E.; Renkens, I.; Duran, K.; Ballarati, L.; Vergult, S.; Giardino, D.; Hansson, K.; et al. Constitutional Chromothripsis Rearrangements Involve Clustered Double-Stranded DNA Breaks and Nonhomologous Repair Mechanisms. Cell Rep. 2012, 1, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; PCAWG Structural Variation Working Group; Lee, J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.A.; Klimczak, L.J.; Zhang, C.-Z.; et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.Y.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018, 37, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, A.; Phadke, T.; Barrera, L.A.; Margulies, C.M.; Lee, C.S.; Buquicchio, F.; Moss, S.; Abdulkerim, H.S.; Selleck, W.; Jayaram, H.; et al. Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun. 2017, 8, 13905. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer/Disease | Affected Repair Pathway | Genetic Background | Drugable Targets | Effects on Genome | References |
|---|---|---|---|---|---|
| Breast, ovarian, melanoma, prostate, pancreatic | HR | BRCA1 deficient | POLQ, PARP1 | Translocations, TDs, LOH, point mutations | [117] |
| Breast, ovarian | HR | BRCA2 deficient | POLQ, PARP1 | Translocations, LOH, TDs, point mutations | [41] |
| Epithelial ovarian cancers | HR | FANCD2 deficient, Increased expression of POLQ | PARP1, FANCD2, POLQ | Chromosomal aberrations, nonsynonymous mutations | [118] |
| Breast, ovarian, Fanconi anemia | HR | PALB2 deficient | PARP1 | Translocations, LOH, TDs, point mutations | [117] |
| Breast, stomach, prostate | HR | CHD1 deficient, Deficient in CtIP recruitment to DSBs | PARP1, PTEN | Translocations, LOH, TDs, point mutations | [119] |
| Breast, ovarian, Fanconi anemia | HR | RAD51C deficient | PARP1 | Genomic instability, aneuploidy, chromosome aberrations | [120] |
| Breast, ovarian, Fanconi anemia | HR | RAD51D deficient | PARP1 | Large deletions, genomic instability | [121] |
| Chronic myeloid leukemia | HR | BCR-ABL (constitutively active tyrosine kinase), increased expression of LIG3a, PARP1, and WRN | PARP1 combined with DNA ligase inhibitors | Translocations, LOH, TDs, point mutations | [122,123] |
| Lynch Syndrome, colorectal, endometrial, ovarian, gastric, urinary, small bowel, pancreatic, prostate | MMR, HR | MLH1, MSH2, MSH6, PMS2 deficient | PARP1 | Point mutations, microsatellite instability | [124] |
| High grade bladder tumors, colon | cNHEJ | KU, DNA-PK, or XRCC4 deficient | Unknown | Deletions (<125 bp on average) and microhomology at repair junctions | [125] |
| Multiple myeloma, leukemia, pro B-cell lymphoma | cNHEJ | KU and P53 deficient | Unknown | Rearrangements and gene amplifications, nonreciprocal translocations, increased microhomologies at repair junctions | [126,127,128] |
| MCF7 breast cancer | cNHEJ | Reduced levels of DNA LIG4 Increased levels of DNA LIG3a and PARP1 | PARP1 combined with DNA ligase inhibitors | Large deletions, translocations | [129] |
| Acute myeloid leukemia | cNHEJ | Decreased expression of KU proteins Increased expression of DNA LIG3a | Unknown | TDs, microhomology at repair junctions, deletions | [130] |
| Neuroblastoma | cNHEJ | LIG4 and Artemis deficient Increased expression of DNA LIG3, LIG1, and PARP1 | PARP1 | Translocations, TDs | [131] |
| Non-BRCA1/2 breast cancer | cNHEJ | XRCC4 deficient | Unknown | Translocations | [132] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanscom, T.; McVey, M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells 2020, 9, 1657. https://doi.org/10.3390/cells9071657
Hanscom T, McVey M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells. 2020; 9(7):1657. https://doi.org/10.3390/cells9071657
Chicago/Turabian StyleHanscom, Terrence, and Mitch McVey. 2020. "Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution" Cells 9, no. 7: 1657. https://doi.org/10.3390/cells9071657
APA StyleHanscom, T., & McVey, M. (2020). Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells, 9(7), 1657. https://doi.org/10.3390/cells9071657