Opposing Roles of S1P3 Receptors in Myocardial Function

 ,
,  , ,
, ,  and
and 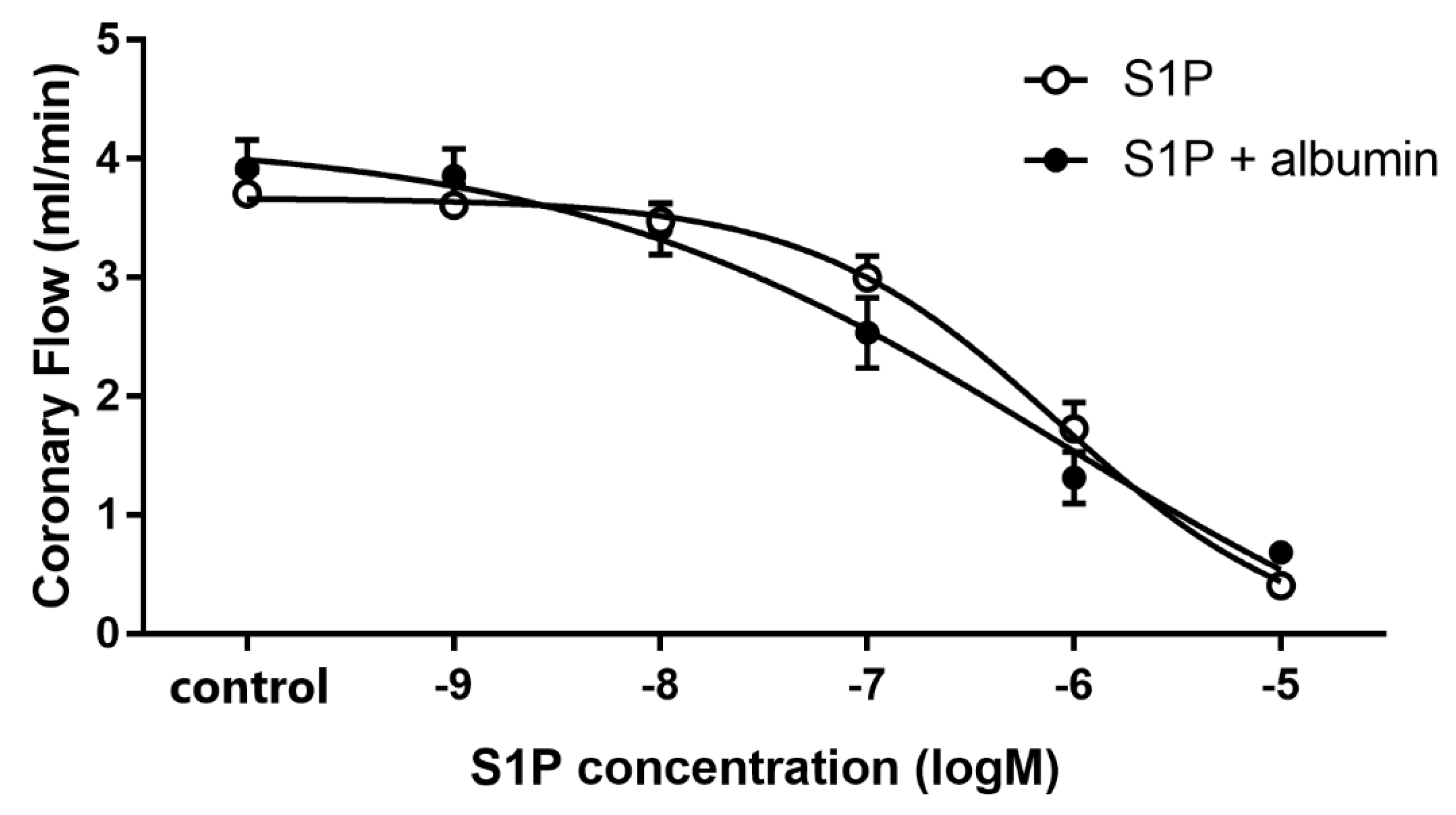{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolated Perfused Heart Experiments
2.3. Experimental Protocol
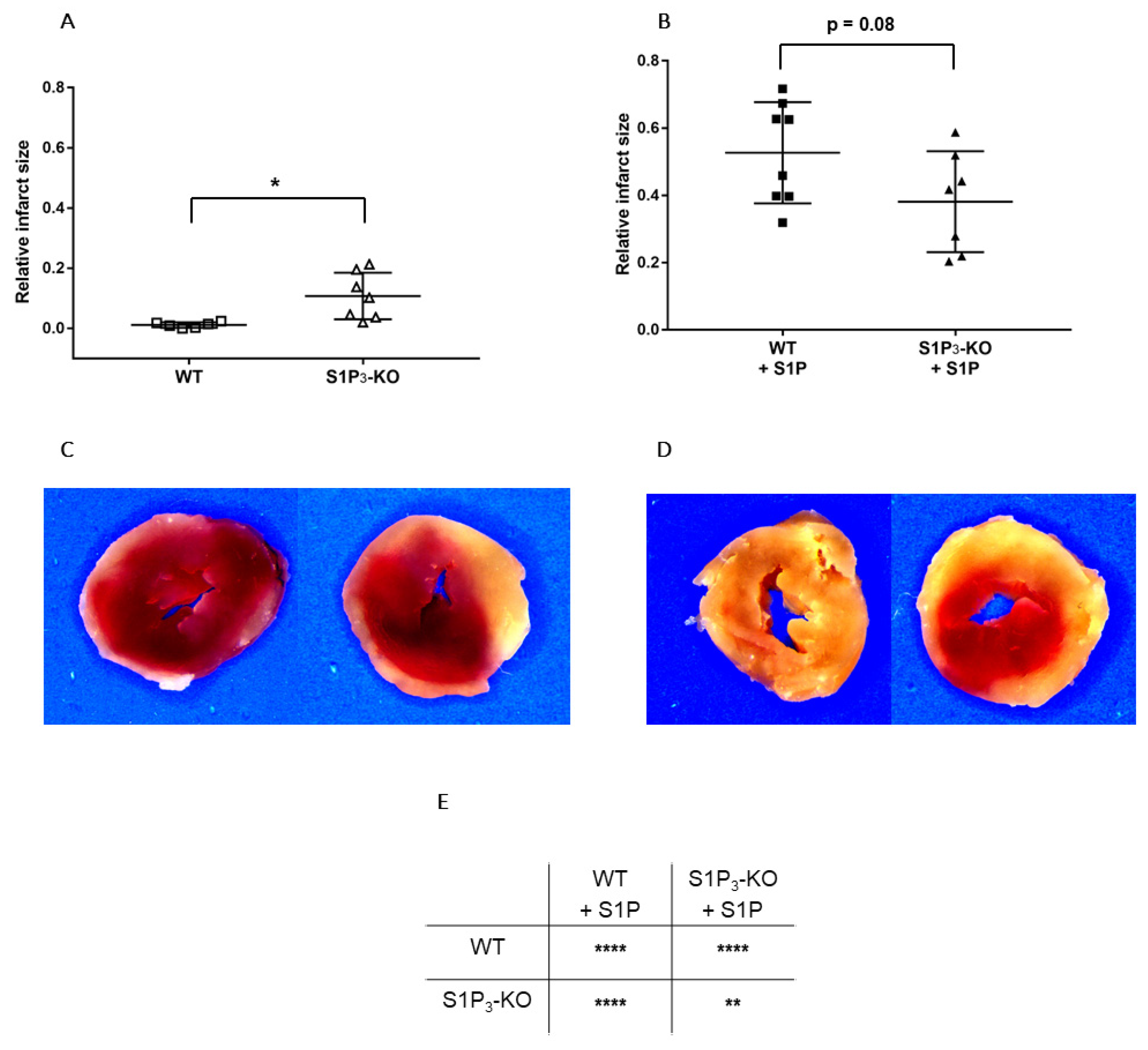
2.4. Measurement of Infarct Size
2.5. Statistical Analysis
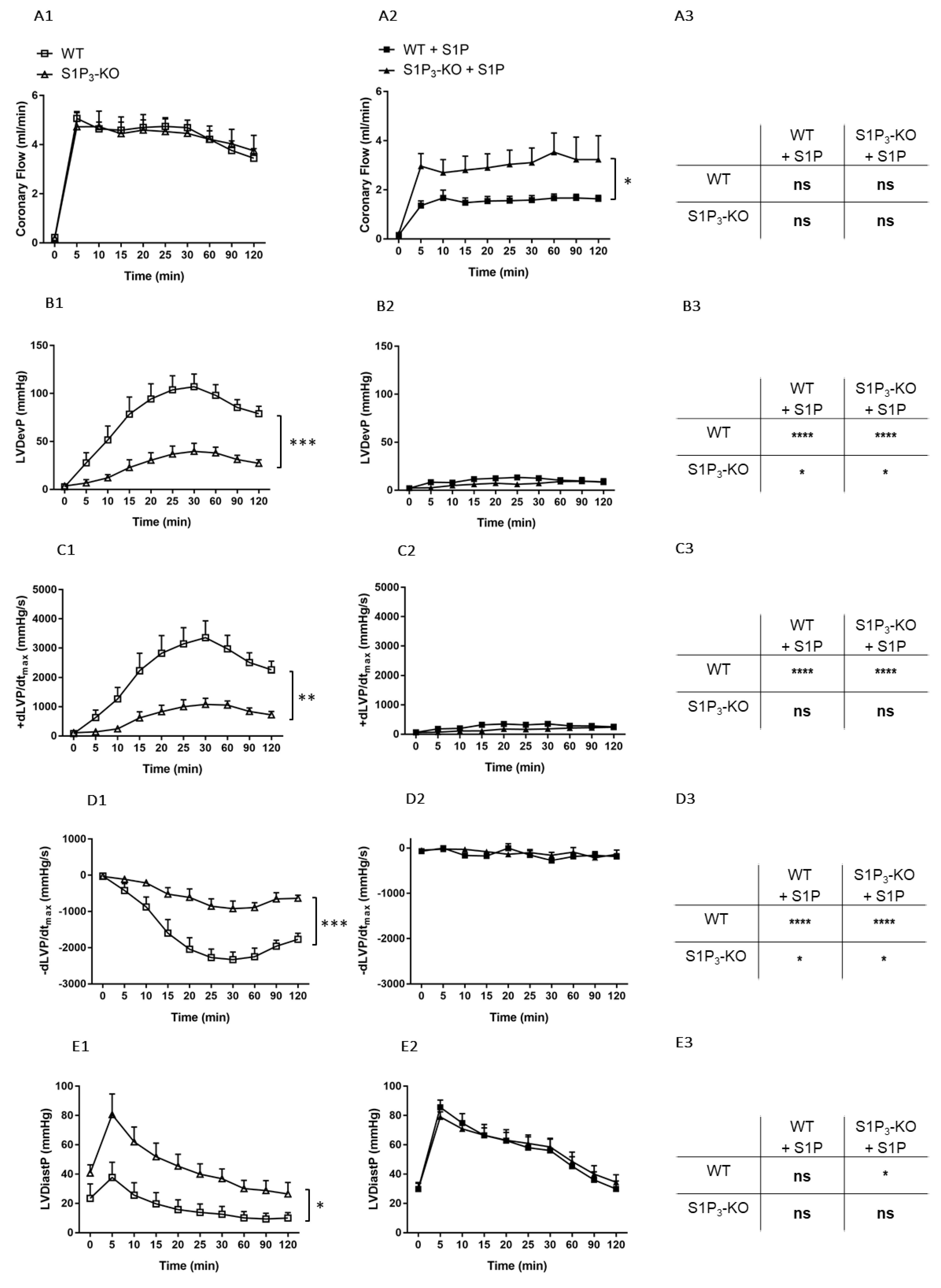
3. Results
3.1. Dose-Dependent Effects of Intravascular S1P on CF Administered with or without S1P-Chaperon Albumin
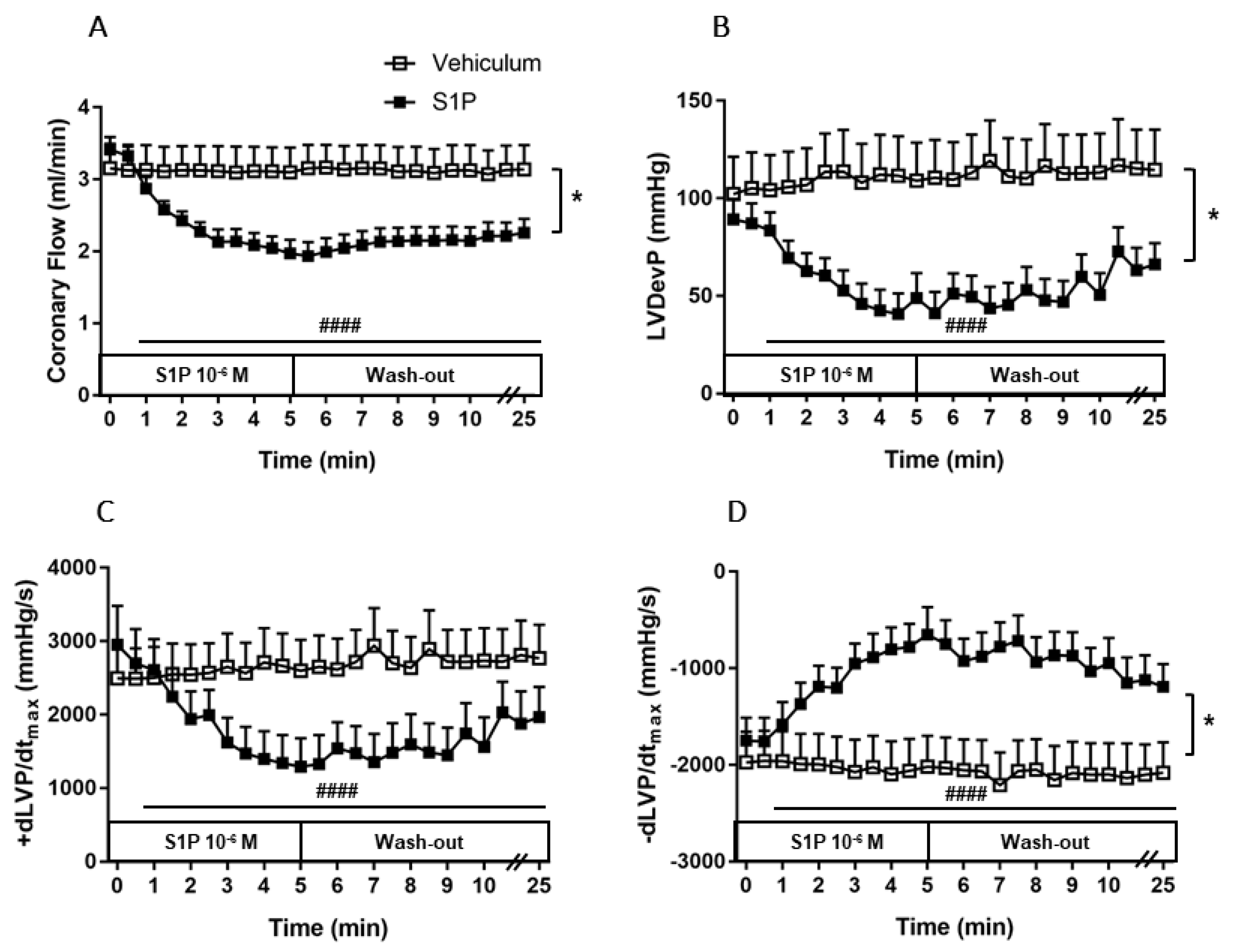
3.2. Effects of Intravascular S1P Exposition on CF and Heart Function
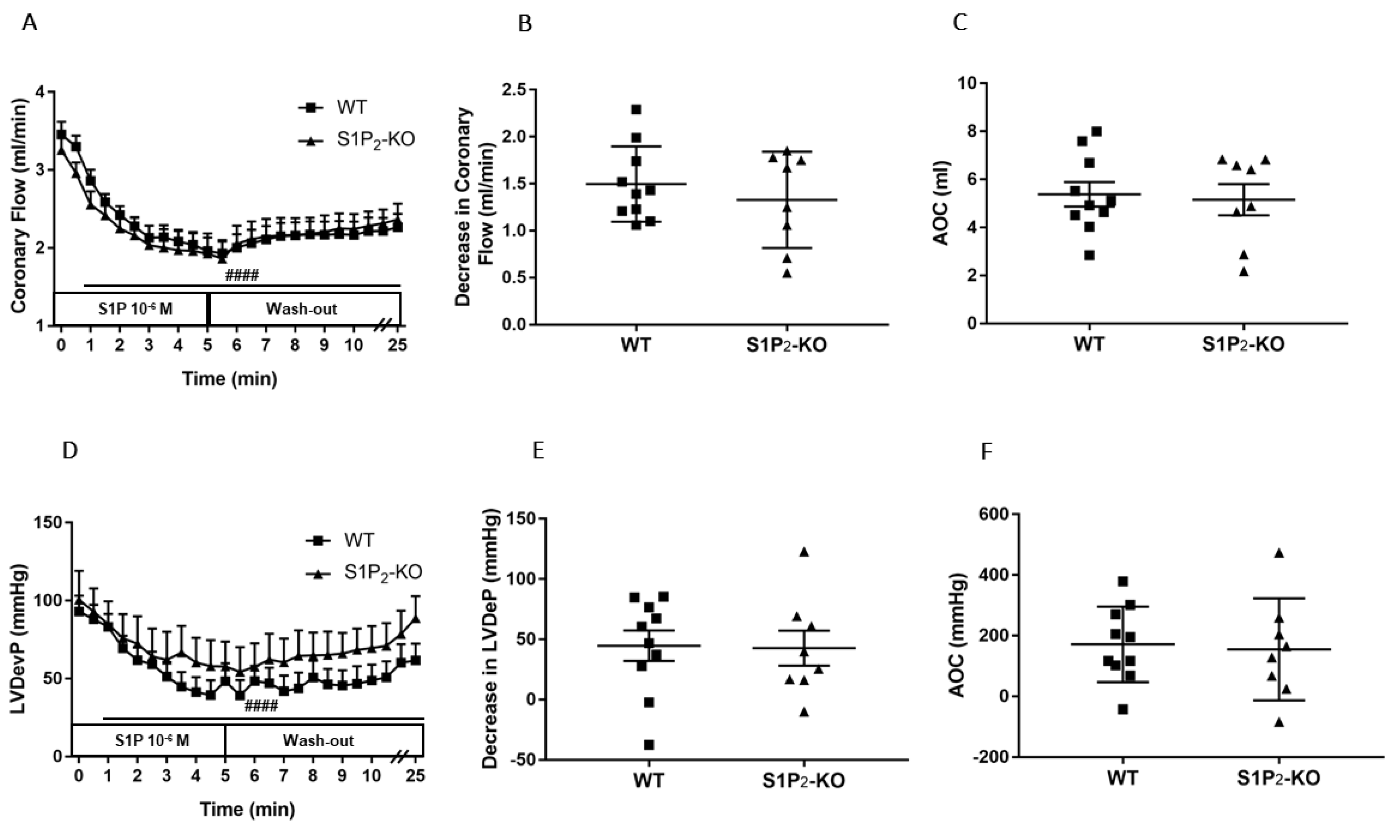
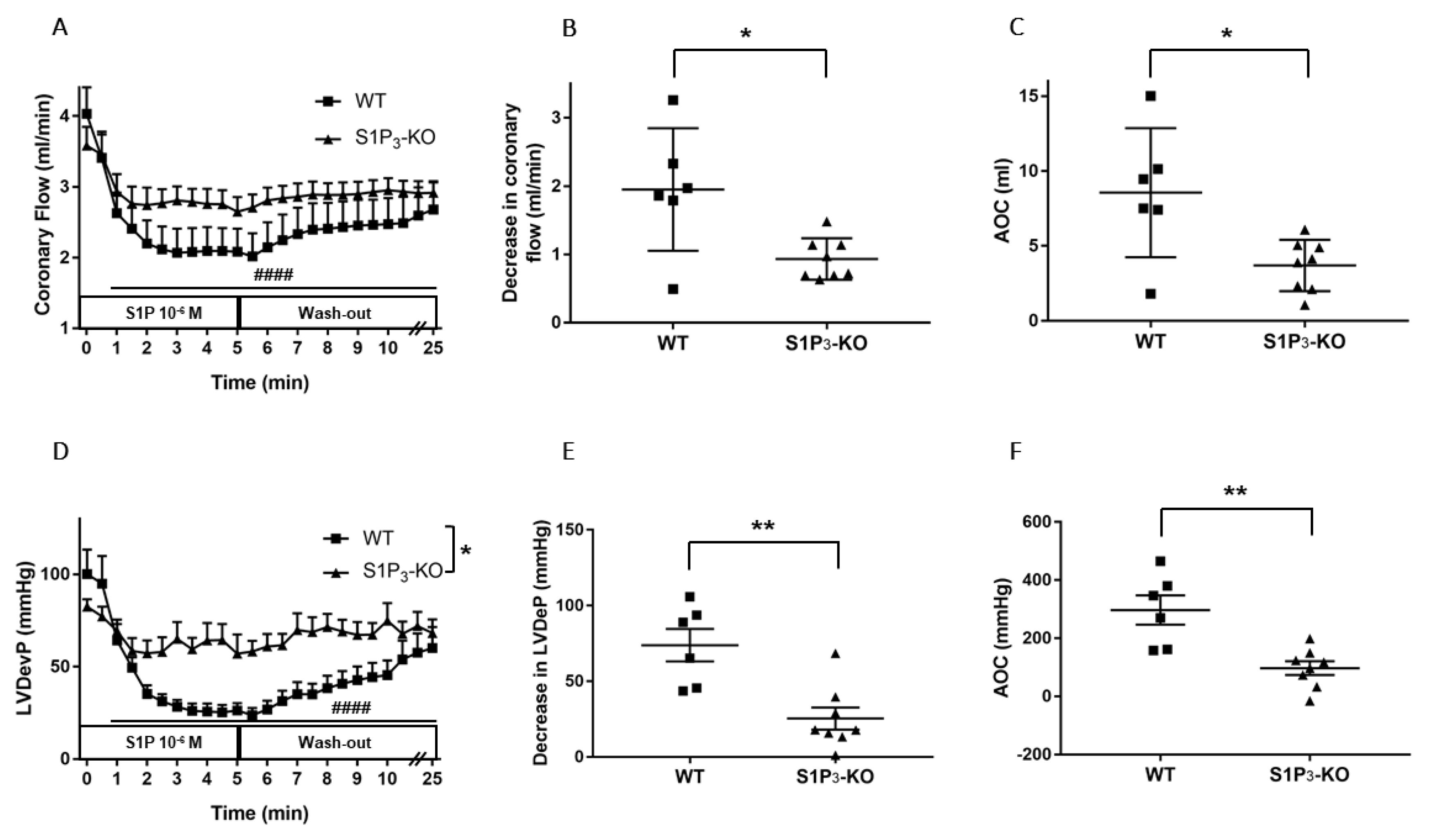
3.3. Role of Myocardial S1P3 Receptor Activation in I/R Injury
3.4. Effects of Preischemic Intravascular S1P Exposure on I/R Injury
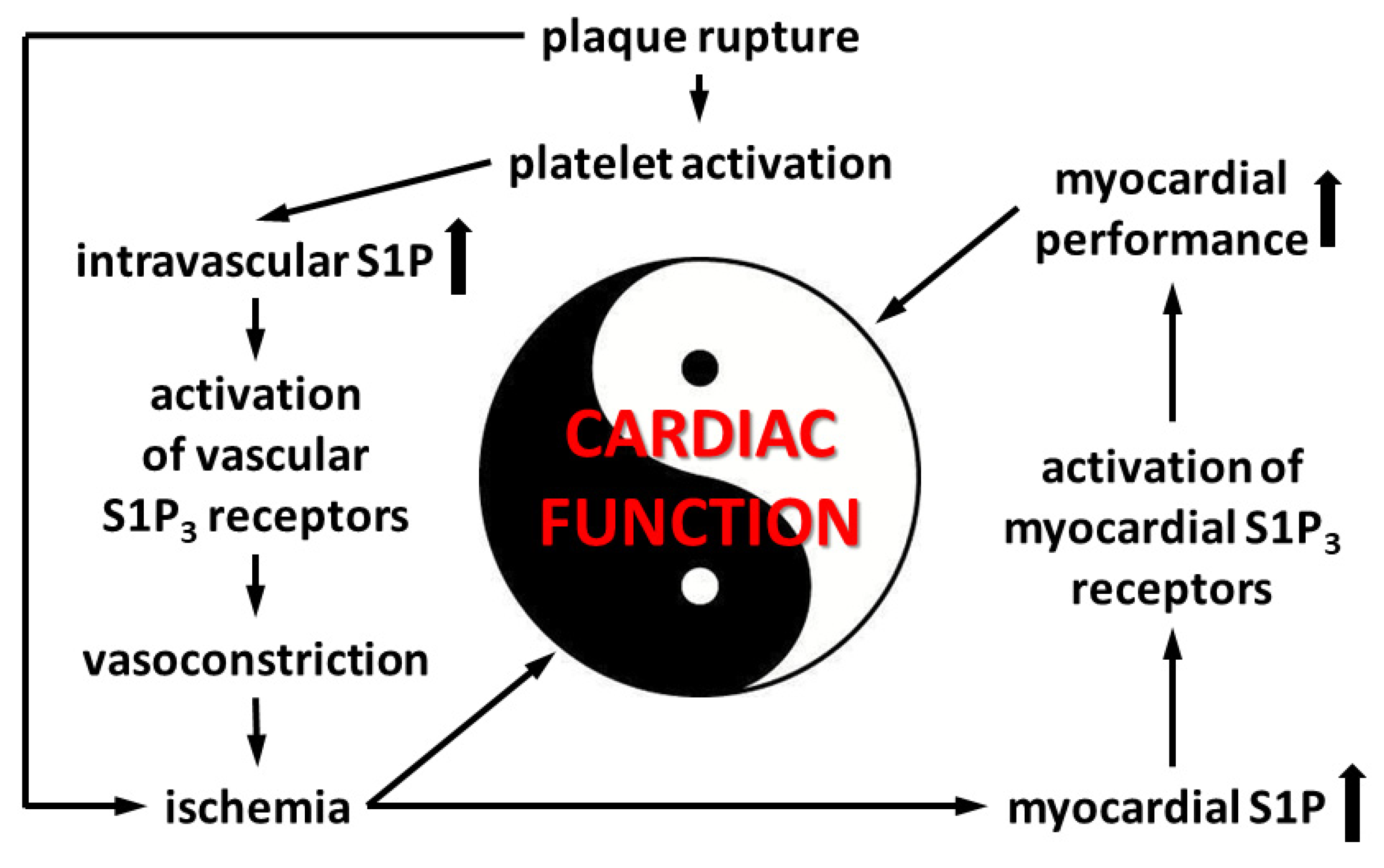
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 2013, 383, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Makki, N.; Brennan, T.M.; Girotra, S. Acute coronary syndrome. J. Intensive Care Med. 2015, 30, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nüsing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schror, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011, 9, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Sano, T.; Ito, M.; Igarashi, Y.; Moschetta, A.; Xu, F.; Hagey, L.R.; Van Berge-Henegouwen, G.P.; Van Erpecum, K.J.; Brouwers, J.F.; et al. Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J. Lipid Res. 2005, 46, 2458–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatomi, Y.; Igarashi, Y.; Yang, L.; Hisano, N.; Qi, R.; Asazuma, N.; Satoh, K.; Ozaki, Y.; Kume, S. Sphingosine 1-phosphate, a bioactive sphingolipid abundantly stored in platelets, is a normal constituent of human plasma and serum. J. Biochem. 1997, 121, 969–973. [Google Scholar] [CrossRef]
- Ono, Y.; Kurano, M.; Ohkawa, R.; Yokota, H.; Igarashi, K.; Aoki, J.; Tozuka, M.; Yatomi, Y. Sphingosine 1-phosphate release from platelets during clot formation: Close correlation between platelet count and serum sphingosine 1-phosphate concentration. Lipids Heal. Dis. 2013, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Yatomi, Y.; Ohmori, T.; Rile, G.; Kazama, F.; Okamoto, H.; Sano, T.; Satoh, K.; Kume, S.; Tigyi, G.; Igarashi, Y.; et al. Sphingosine 1-phosphate as a major bioactive lysophospholipid that is released from platelets and interacts with endothelial cells. Blood 2000, 96, 3431–3438. [Google Scholar] [CrossRef]
- Yatomi, Y.; Ruan, F.; Hakomori, S.; Igarashi, Y. Sphingosine-1-phosphate: A platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood 1995, 86, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular Endothelium As a Contributor of Plasma Sphingosine 1-Phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Blaho, V.A.; Galvani, S.; Engelbrecht, E.; Liu, C.; Swendeman, S.L.; Kono, M.; Proia, R.L.; Steinman, L.; Han, M.H.; Hla, T. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 2015, 523, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, M.; Okada, H.; Dahlbäck, B. HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Heal. Dis. 2017, 16, 36. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-C.; Han, M.H. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway: Therapeutic Targets in Autoimmunity and Inflammation. Drugs 2016, 76, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Bieberich, E. Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv. Boil. Regul. 2018, 70, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Michel, T. Sphingosine-1-phosphate and modulation of vascular tone. Cardiovasc. Res. 2009, 82, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerage, D.; Brindley, D.; Hemmings, D.G. Review: Novel insights into the regulation of vascular tone by sphingosine 1-phosphate. Placenta 2014, 35, S86–S92. [Google Scholar] [CrossRef]
- Peters, S.L.; Alewijnse, A. Sphingosine-1-phosphate signaling in the cardiovascular system. Curr. Opin. Pharmacol. 2007, 7, 186–192. [Google Scholar] [CrossRef]
- Siess, W. Athero- and thrombogenic actions of lysophosphatidic acid and sphingosine-1-phosphate. Biochim. Et Biophys. Acta (Bba) Bioenerg. 2002, 1582, 204–215. [Google Scholar] [CrossRef]
- Karliner, J.S. Sphingosine kinase and sphingosine 1-phosphate in the heart: A decade of progress. Biochim. Et Biophys. Acta (Bba) Mol. Cell Boil. Lipids 2013, 1831, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Mutoh, T.; Rivera, R.; Chun, J. Insights into the pharmacological relevance of lysophospholipid receptors. Br. J. Pharmacol. 2012, 165, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Tigyi, G.J. Lysophospholipid Receptors. In Encyclopedia of Biological Chemistry, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 1–3. [Google Scholar]
- Xiang, S.Y.; Ouyang, K.; Yung, B.S.; Miyamoto, S.; Smrcka, A.V.; Chen, J.; Brown, J.H. PLC, PKD1, and SSH1L Transduce RhoA Signaling to Protect Mitochondria from Oxidative Stress in the Heart. Sci. Signal. 2013, 6, ra108. [Google Scholar] [CrossRef] [Green Version]
- Karliner, J.S.; Honbo, N.; Summers, K.; Gray, M.O.; Goetzl, E.J. The Lysophospholipids Sphingosine-1-Phosphate and Lysophosphatidic Acid Enhance Survival during Hypoxia in Neonatal Rat Cardiac Myocytes. J. Mol. Cell. Cardiol. 2001, 33, 1713–1717. [Google Scholar] [CrossRef]
- Jin, Z.-Q.; Goetzl, E.J.; Karliner, J.S. Sphingosine Kinase Activation Mediates Ischemic Preconditioning in Murine Heart. Circulation 2004, 110, 1980–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessey, D.A.; Kelley, M.; Li, L.; Huang, Y.; Zhou, H.-Z.; Zhu, B.Q.; Karliner, J.S. Role of sphingosine kinase activity in protection of heart against ischemia reperfusion injury. Med. Sci. Monit. 2006, 12, 318–324. [Google Scholar]
- Means, C.K.; Xiao, C.-Y.; Li, Z.; Zhang, T.; Omens, J.H.; Ishii, I.; Chun, J.; Brown, J.H. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2007, 292, H2944–H2951. [Google Scholar] [CrossRef] [PubMed]
- Yung, B.S.; Brand, C.S.; Xiang, S.Y.; Gray, C.B.B.; Means, C.K.; Rosen, H.; Chun, J.; Purcell, N.H.; Brown, J.H.; Miyamoto, S. Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J. Mol. Cell. Cardiol. 2016, 103, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theilmeier, G.; Schmidt, C.; Herrmann, J.; Keul, P.; Schäfers, M.; Herrgott, I.; Mersmann, J.; Larmann, J.; Hermann, S.; Stypmann, J.; et al. High-Density Lipoproteins and Their Constituent, Sphingosine-1-Phosphate, Directly Protect the Heart Against Ischemia/Reperfusion Injury In Vivo via the S1P 3 Lysophospholipid Receptor. Circulation 2006, 114, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, S.-H.; Constable, P.D.; Smith, G.W.; Haschek, W.M. Effects of Exogenous Sphinganine, Sphingosine, and Sphingosine-1-Phosphate on Relaxation and Contraction of Porcine Thoracic Aortic and Pulmonary Arterial Rings. Toxicol. Sci. 2005, 86, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Tosaka, M.; Okajima, F.; Hashiba, Y.; Saito, N.; Nagano, T.; Watanabe, T.; Kimura, T.; Sasaki, T. Sphingosine 1-phosphate contracts canine basilar arteries in vitro and in vivo: Possible role in pathogenesis of cerebral vasospasm. Stroke 2001, 32, 2913–2919. [Google Scholar] [CrossRef] [Green Version]
- Salomone, S.; Yoshimura, S.-I.; Reuter, U.; Foley, M.; Thomas, S.S.; Moskowitz, M.A.; Waeber, C. S1P3 receptors mediate the potent constriction of cerebral arteries by sphingosine-1-phosphate. Eur. J. Pharmacol. 2003, 469, 125–134. [Google Scholar] [CrossRef]
- Ikeda, H.; Nagashima, K.; Yanase, M.; Tomiya, T.; Arai, M.; Inoue, Y.; Tejima, K.; Nishikawa, T.; Watanabe, N.; Omata, M.; et al. Sphingosine 1-phosphate enhances portal pressure in isolated perfused liver via S1P2 with Rho activation. Biochem. Biophys. Res. Commun. 2004, 320, 754–759. [Google Scholar] [CrossRef]
- Sugiyama, A.; Yatomi, Y.; Ozaki, Y.; Hashimoto, K. Sphingosine 1-phosphate induces sinus tachycardia and coronary vasoconstriction in the canine heart. Cardiovasc. Res. 2000, 46, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-Phosphate (S1P) Regulates Vascular Contraction via S1P3 Receptor: Investigation Based on a New S1P3 Receptor Antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, T.; Yatomi, Y.; Osada, M.; Kazama, F.; Takafuta, T.; Ikeda, H.; Ozaki, Y. Sphingosine 1-phosphate induces contraction of coronary artery smooth muscle cells via S1P. Cardiovasc. Res. 2003, 58, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Means, C.K.; Miyamoto, S.; Chun, J.; Brown, J.H. S1P1 receptor localization confers selectivity for G i-mediated cAMP and contractile responses. J. Biol. Chem. 2008, 283, 11954–11963. [Google Scholar] [CrossRef] [Green Version]
- Landeen, L.K.; Dederko, D.A.; Kondo, C.S.; Hu, B.S.; Aroonsakool, N.; Haga, J.H.; Giles, W.R. Mechanisms of the negative inotropic effects of sphingosine-1-phosphate on adult mouse ventricular myocytes. Am. J. Physiol. Circ. Physiol. 2008, 294, H736–H749. [Google Scholar] [CrossRef] [PubMed]
- Sanna, M.G.; Liao, J.; Jo, E.; Alfonso, C.; Ahn, M.-Y.; Peterson, M.S.; Webb, B.; Lefebvre, S.; Chun, J.; Gray, N.; et al. Sphingosine 1-Phosphate (S1P) Receptor Subtypes S1P1and S1P3, Respectively, Regulate Lymphocyte Recirculation and Heart Rate. J. Boil. Chem. 2004, 279, 13839–13848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, M.; Sun, S.-Y.; Hajdu, R.; Bergstrom, J.; Card, D.; Doherty, G.; Hale, J.; Keohane, C.; Meyers, C.; Milligan, J.; et al. Immune Cell Regulation and Cardiovascular Effects of Sphingosine 1-Phosphate Receptor Agonists in Rodents Are Mediated via Distinct Receptor Subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, M.; Mi, Y.; Liu, Y.; Sasaki, T.; Allende, M.L.; Wu, Y.-P.; Yamashita, T.; Proia, R.L. The Sphingosine-1-phosphate Receptors S1P1, S1P2, and S1P3Function Coordinately during Embryonic Angiogenesis. J. Boil. Chem. 2004, 279, 29367–29373. [Google Scholar] [CrossRef] [Green Version]
- Kemecsei, P.; Miklós, Z.; Bíró, T.; Marincsák, R.; Tóth, B.I.; Komlodi-Pasztor, E.; Barnucz, E.; Mirk, E.; Van Der Vusse, G.J.; Ligeti, L.; et al. Hearts of surviving MLP-KO mice show transient changes of intracellular calcium handling. Mol. Cell. Biochem. 2010, 342, 251–260. [Google Scholar] [CrossRef]
- Miklós, Z.; Kemecsei, P.; Biro, T.; Marincsak, R.; Tóth, B.I.; Buijs, J.; Benis, E.; Drozgyik, A.; Ivanics, T. Early cardiac dysfunction is rescued by upregulation of SERCA2a pump activity in a rat model of metabolic syndrome. Acta Physiol. 2012, 205, 381–393. [Google Scholar] [CrossRef]
- Skrzypiec-Spring, M.; Grotthus, B.; Szelag, A.; Schulz, R. Isolated heart perfusion according to Langendorff—Still viable in the new millennium. J. Pharmacol. Toxicol. Methods 2007, 55, 113–126. [Google Scholar] [CrossRef]
- Lacza, Z.; Dézsi, L.; Káldi, K.; Horvath, E.M.; Sándor, P.; Benyó, Z. Prostacyclin-mediated compensatory mechanism in the coronary circulation during acute NO synthase blockade. Life Sci. 2003, 73, 1141–1149. [Google Scholar] [CrossRef]
- Liliom, K.; Sun, G.; Bünemann, M.; Virág, T.; Nusser, N.; Baker, D.L.; Wang, D.A.; Fabian, M.J.; Brandts, B.; Bender, K.; et al. Sphingosylphosphocholine is a naturally occurring lipid mediator in blood plasma: A possible role in regulating cardiac function via sphingolipid receptors. Biochem. J. 2001, 355, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Nachlas, M.M.; Shnitka, T.K. Macroscopic Identification of Early Myocardial Infarcts by Alterations in Dehydrogenase Activity. Am. J. Pathol. 1963, 42, 379–405. [Google Scholar]
- Hemmings, D.G.; Xu, Y.; Davidge, S.T. Sphingosine 1-phosphate-induced vasoconstriction is elevated in mesenteric resistance arteries from aged female rats. Br. J. Pharmacol. 2004, 143, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmings, D.G.; Hudson, N.K.; Halliday, D.; O’Hara, M.; Baker, P.N.; Davidge, S.T.; Taggart, M. Sphingosine-1-Phosphate Acts via Rho-Associated Kinase and Nitric Oxide to Regulate Human Placental Vascular Tone1. Boil. Reprod. 2006, 74, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, B.A.; Argraves, K.M. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim. Et Biophys. Acta (Bba) Mol. Cell Boil. Lipids 2014, 1841, 1403–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Ruiz, M.; Lee, M.-J.; Zöllner, S.; Gratton, J.-P.; Scotland, R.; Shiojima, I.; Walsh, K.; Hla, T.; Sessa, W.C. Sphingosine 1-Phosphate Activates Akt, Nitric Oxide Production, and Chemotaxis through a GiProtein/Phosphoinositide 3-Kinase Pathway in Endothelial Cells. J. Boil. Chem. 2001, 276, 19672–19677. [Google Scholar] [CrossRef] [Green Version]
- Nofer, J.-R.; Van Der Giet, M.; Tolle, M.; Wolinska, I.; Lipinski, K.V.W.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef]
- Levkau, B.; Hermann, S.; Theilmeier, G.; Van Der Giet, M.; Chun, J.; Schober, O.; Schäfers, M. High-Density Lipoprotein Stimulates Myocardial Perfusion In Vivo. Circulation 2004, 110, 3355–3359. [Google Scholar] [CrossRef] [Green Version]
- Deutschman, D.H.; Carstens, J.S.; Klepper, R.L.; Smith, W.S.; Page, M.; Young, T.R.; Gleason, L.A.; Nakajima, N.; Sabbadini, R.A. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Hear. J. 2003, 146, 62–68. [Google Scholar] [CrossRef]
- Okajima, F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: Is this an atherogenic mediator or an anti-atherogenic mediator? Biochim. Et Biophys. Acta (Bba) Bioenerg. 2002, 1582, 132–137. [Google Scholar] [CrossRef]
- Panta, C.R.; Ruisanchez, É.; Móré, D.; Dancs, P.T.; Balogh, A.; Fülöp, Á.; Kerék, M.; Proia, R.L. Sphingosine-1-phosphate enhances α1-adrenergic vasoconstriction via S1P2–G12/13–ROCK mediated signaling. Int. J. Mol. Sci. 2019, 20, 6361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karliner, J.S. Sphingosine Kinase and Sphingosine 1-Phosphate in Cardioprotection. J. Cardiovasc. Pharmacol. 2009, 53, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, M. Cardioprotective role of sphingosine-1-phosphate. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2011, 62, 601–607. [Google Scholar]
- Xin, C.; Ren, S.; Pfeilschifter, J.; Huwiler, A. Heterologous desensitization of the sphingosine-1-phosphate receptors by purinoceptor activation in renal mesangial cells. Br. J. Pharmacol. 2004, 143, 581–589. [Google Scholar] [CrossRef]
- Gräler, M.H.; Goetzl, E.J. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G protein-coupled receptors. Faseb J. 2004, 18, 551–553. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wafa, D.; Koch, N.; Kovács, J.; Kerék, M.; Proia, R.L.; Tigyi, G.J.; Benyó, Z.; Miklós, Z. Opposing Roles of S1P3 Receptors in Myocardial Function. Cells 2020, 9, 1770. https://doi.org/10.3390/cells9081770
Wafa D, Koch N, Kovács J, Kerék M, Proia RL, Tigyi GJ, Benyó Z, Miklós Z. Opposing Roles of S1P3 Receptors in Myocardial Function. Cells. 2020; 9(8):1770. https://doi.org/10.3390/cells9081770
Chicago/Turabian StyleWafa, Dina, Nóra Koch, Janka Kovács, Margit Kerék, Richard L. Proia, Gábor J. Tigyi, Zoltán Benyó, and Zsuzsanna Miklós. 2020. "Opposing Roles of S1P3 Receptors in Myocardial Function" Cells 9, no. 8: 1770. https://doi.org/10.3390/cells9081770
APA StyleWafa, D., Koch, N., Kovács, J., Kerék, M., Proia, R. L., Tigyi, G. J., Benyó, Z., & Miklós, Z. (2020). Opposing Roles of S1P3 Receptors in Myocardial Function. Cells, 9(8), 1770. https://doi.org/10.3390/cells9081770