Sphingolipids in Type 1 Diabetes: Focus on Beta-Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
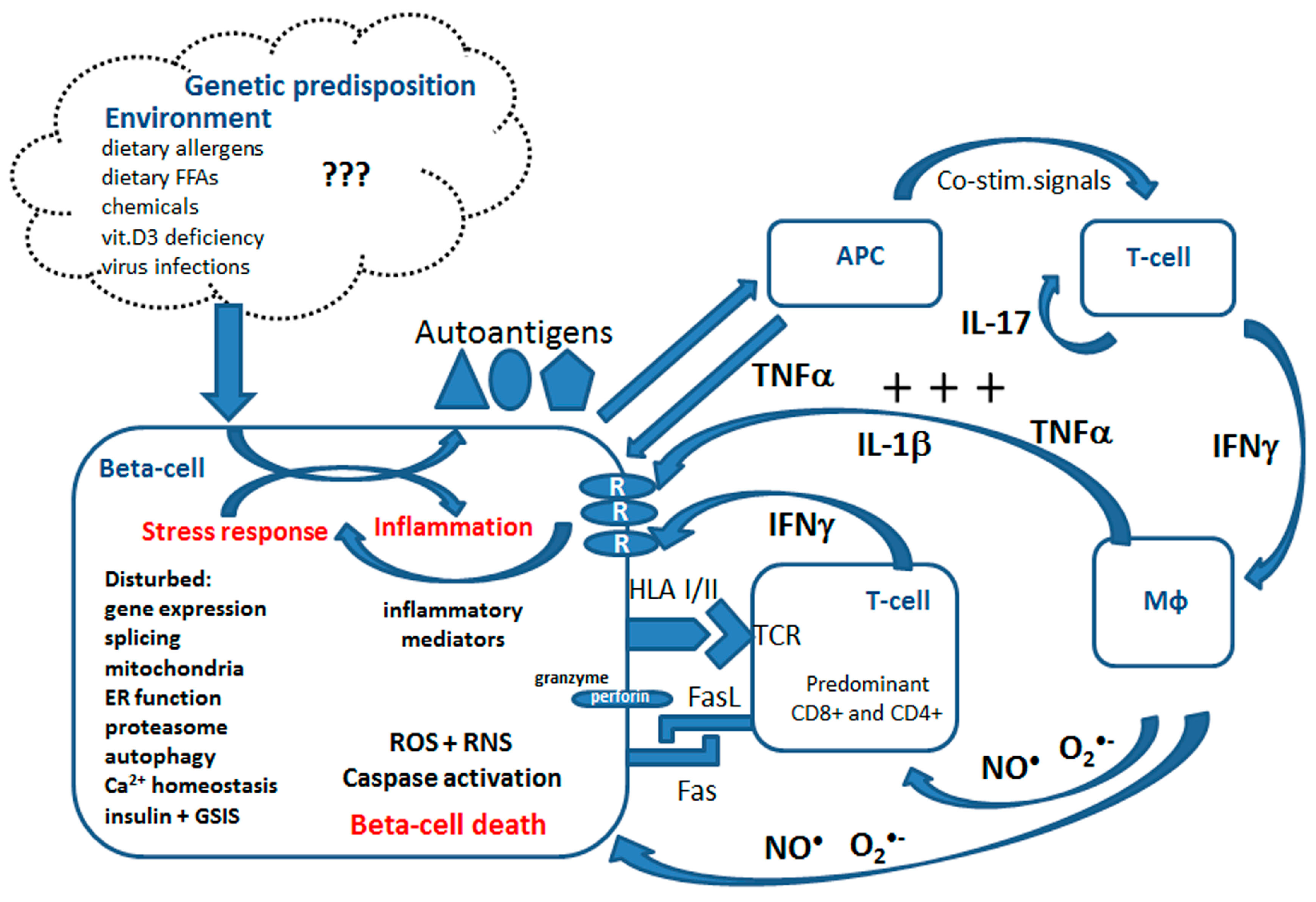
2. Overview of Mechanisms of Beta-Cell Destruction in T1DM
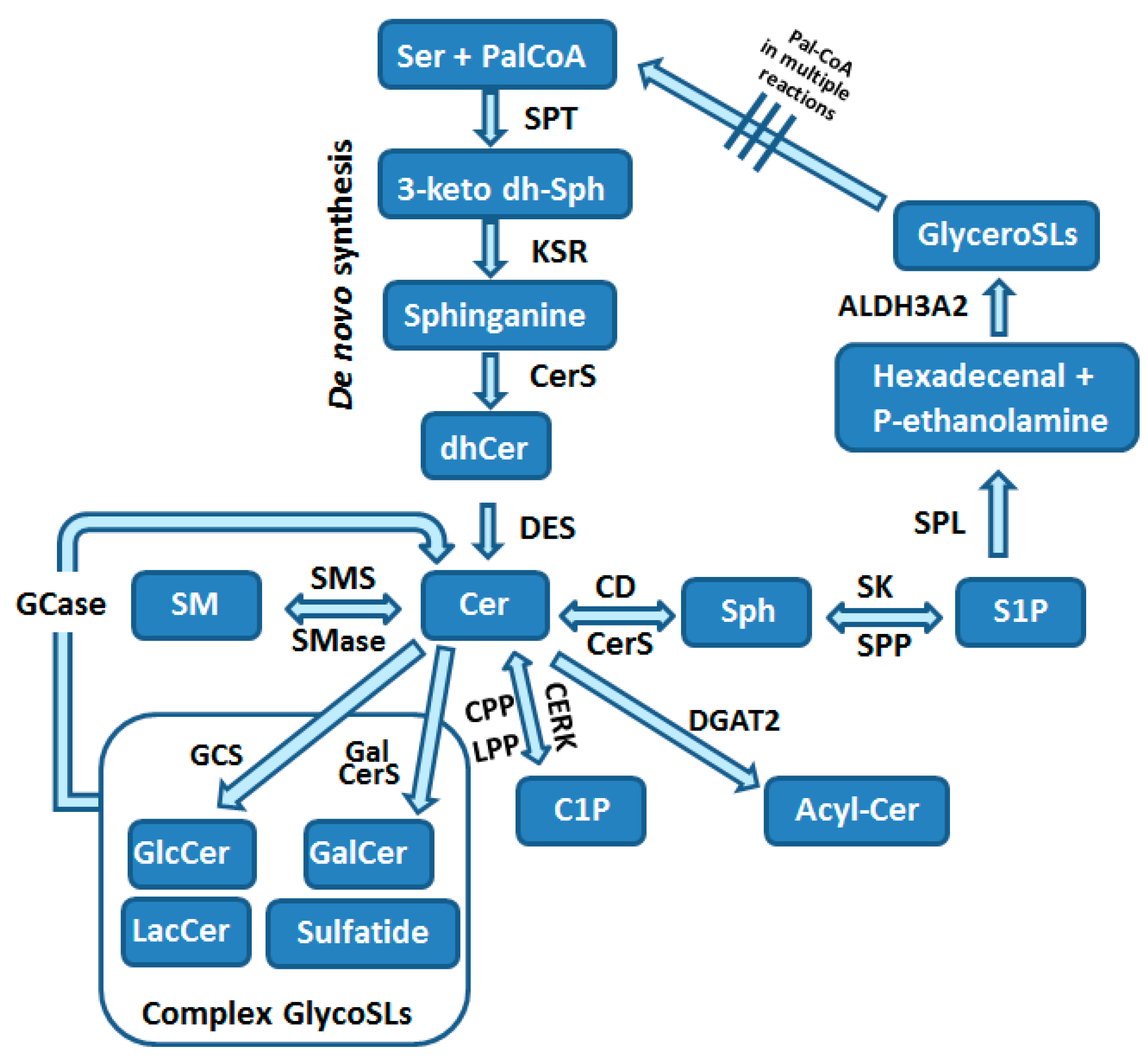
3. Biosynthesis of Sphingolipids
4. Overview of Major Functions of Bioactive Sphingolipids
4.1. Ceramide
4.2. Sphingosine
4.3. Sphingosine 1 Phosphate
4.4. Ceramide-1 Phosphate
4.5. Sulfatide
5. Effects of Proinflammatory Cytokines on the Sphingolipid Pathway Enzymes in Pancreatic Beta-Cells
5.1. De Novo Synthesis
5.2. Ceramide Metabolism Regulation
5.3. S1P Metabolism, Receptors and Transporters
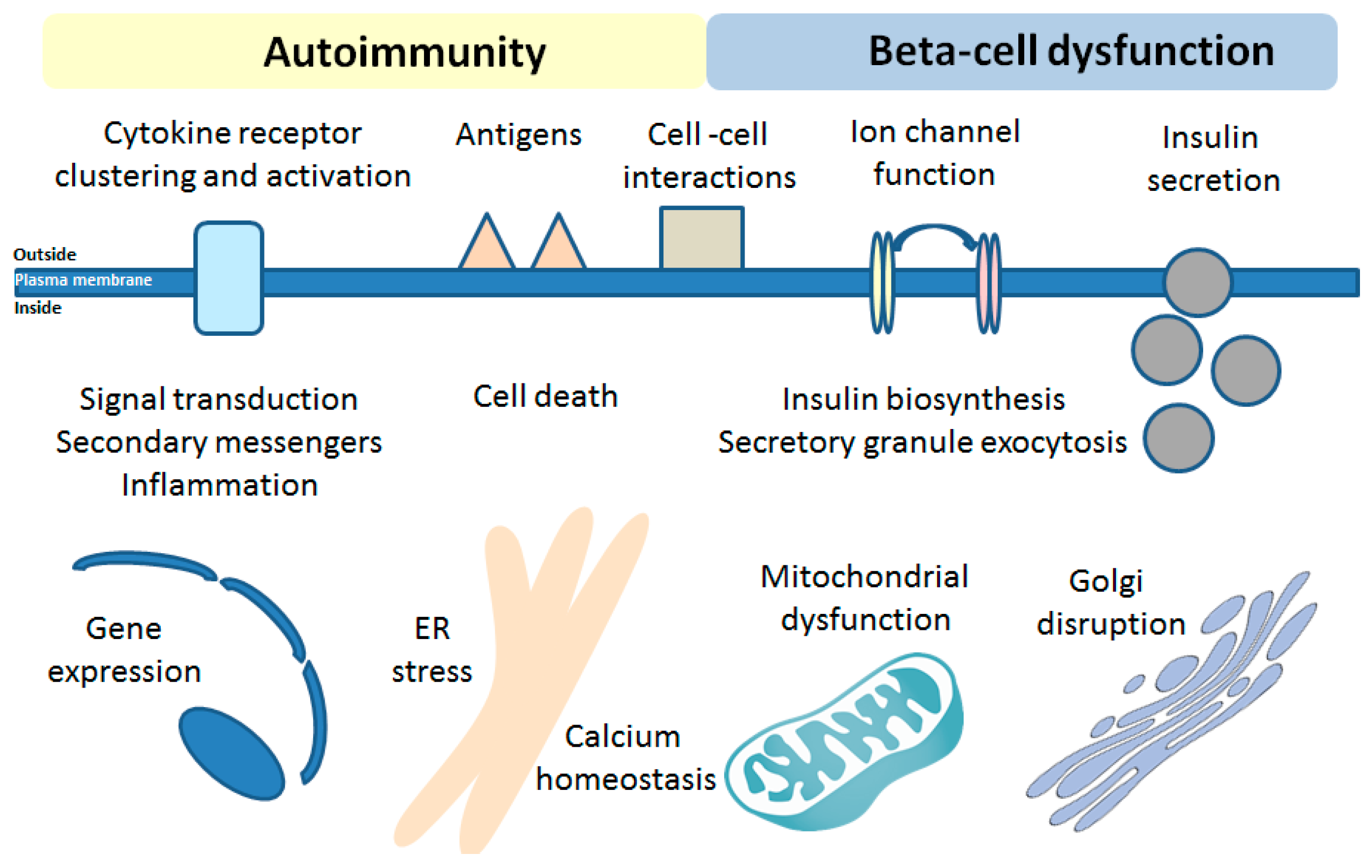
6. Effects of Bioactive Sphingolipids on the Beta-Cell Function in T1DM
7. Effects of Bioactive Sphingolipids on Beta-Cell Fate in T1DM
8. Sphingolipids in Animal Models of Autoimmune Diabetes and Human T1DM
8.1. Animal Models
8.2. Human T1DM
9. Sphingolipids as Autoantigens and Biomarkers in T1DM
10. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trend Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Hla, T.; Dannenberg, A.J. Sphingolipid signaling in metabolic disorders. Cell Metab. 2012, 16, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [Green Version]
- Boslem, E.; MacIntosh, G.; Preston, A.M.; Bartley, C.; Busch, A.K.; Fuller, M.; Laybutt, D.R.; Meikle, P.J.; Biden, T.J. A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 2011, 435, 267–276. [Google Scholar] [CrossRef]
- Jessup, C.F.; Bonder, C.S.; Pitson, S.M.; Coates, P.T. The sphingolipid rheostat: A potential target for improving pancreatic islet survival and function. Endocr. Metab. Immun. Disord. Drug Targ. 2011, 11, 262–272. [Google Scholar] [CrossRef]
- Veret, J.; Bellini, L.; Giussani, P.; Ng, C.; Magnan, C.; Le Stunff, H. Roles of sphingolipid metabolism in pancreatic beta-cell dysfunction induced by lipotoxicity. J. Clin. Med. 2014, 3, 646–662. [Google Scholar] [CrossRef] [Green Version]
- Veret, J.; Coant, N.; Berdyshev, E.V.; Skobeleva, A.; Therville, N.; Bailbe, D.; Gorshkova, I.; Natarajan, V.; Portha, B.; Le Stunff, H. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 beta-cells. Biochem. J. 2011, 438, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Veret, J.; Coant, N.; Gorshkova, I.A.; Giussani, P.; Fradet, M.; Riccitelli, E.; Skobeleva, A.; Goya, J.; Kassis, N.; Natarajan, V.; et al. Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 beta-cell survival. BBA 2013, 1831, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Gjoni, E.; Brioschi, L.; Cinque, A.; Coant, N.; Islam, M.N.; Ng, C.K.; Verderio, C.; Magnan, C.; Riboni, L.; Viani, P.; et al. Glucolipotoxicity impairs ceramide flow from the endoplasmic reticulum to the Golgi apparatus in INS-1 beta-cells. PLoS ONE 2014, 9, e110875. [Google Scholar] [CrossRef]
- Dooley, J.; Tian, L.; Schonefeldt, S.; Delghingaro-Augusto, V.; Garcia-Perez, J.E.; Pasciuto, E.; Di Marino, D.; Carr, E.J.; Oskolkov, N.; Lyssenko, V.; et al. Genetic predisposition for beta-cell fragility underlies type 1 and type 2 diabetes. Nat. Genet. 2016, 48, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.J.; Krogvold, L.; Hasselby, J.P.; Kaur, S.; Claessens, L.A.; Russell, M.A.; Mathews, C.E.; Hanssen, K.F.; Morgan, N.G.; Koeleman, B.P.C.; et al. Abnormal islet sphingolipid metabolism in type 1 diabetes. Diabetologia 2018, 61, 1650–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oresic, M.; Simell, S.; Sysi-Aho, M.; Nanto-Salonen, K.; Seppanen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, N.; Pan, J.; Pop-Busui, R.; Othman, A.; Alecu, I.; Hornemann, T.; Eichler, F.S. Altered sphingoid base profiles in type 1 compared to type 2 diabetes. Lipid Health Dis. 2014, 13, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Dickens, A.M.; Lopez-Bascon, M.A.; Lindeman, T.; Kemppainen, E.; Lamichhane, S.; Ronkko, T.; Ilonen, J.; Toppari, J.; Veijola, R.; et al. Metabolic alterations in immune cells associate with progression to type 1 diabetes. Diabetologia 2020, 63, 1017–1031. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, S.; Ahonen, L.; Dyrlund, T.S.; Dickens, A.M.; Siljander, H.; Hyoty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyotylainen, T.; et al. Circulating metabolites in progression to islet autoimmunity and type 1 diabetes. Diabetologia 2019, 62, 2287–2297. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, S.; Kemppainen, E.; Trost, K.; Siljander, H.; Hyoty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyotylainen, T.; Knip, M.; et al. Cord-Blood Lipidome in Progression to Islet Autoimmunity and Type 1 Diabetes. Biomolecules 2019, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.; Tyka, K.; Saba, J.D.; Lenzen, S.; Gurgul-Convey, E. Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 2017, 292, 20292–20304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, A. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Patterson, C.C.; Dahlquist, G.G.; Gyurus, E.; Green, A.; Soltesz, G.; Group, E.S. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005-20, a multicentre prospective registration study. Lancet 2009, 373, 2027–2033. [Google Scholar] [CrossRef]
- Roep, B.O.; Peakman, M. Diabetogenic T lymphocytes in human type 1 diabetes. Curr Opin. Immunol. 2011, 23, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; von Herrath, M.G. The type 1 diabetes signature: Hardwired to trigger inflammation? Diabetes 2014, 63, 3581–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020. [Google Scholar] [CrossRef]
- Mandrup-Poulsen, T. Beta cell death and protection. Ann. N. Y. Acad. Sci. 2003, 1005, 32–42. [Google Scholar] [CrossRef]
- Nerup, J.; Mandrup-Poulsen, T.; Helqvist, S.; Andersen, H.U.; Pociot, F.; Reimers, J.I.; Cuartero, B.G.; Karlsen, A.E.; Bjerre, U.; Lorenzen, T. On the pathogenesis of IDDM. Diabetologia 1994, 37 (Suppl. 2), S82–S89. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Grieco, F.A. On the immense variety and complexity of circumstances conditioning pancreatic beta-cell apoptosis in type 1 diabetes. Diabetes 2012, 61, 1661–1663. [Google Scholar] [CrossRef] [Green Version]
- van Belle, T.L.; Coppieters, K.T.; von Herrath, M.G. Type 1 diabetes: Etiology; immunology; and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.; Krogvold, L.; Zhitomirsky, S.; Swisa, A.; Fischman, M.; Lax, T.; Dahan, T.; Hurvitz, N.; Weinberg-Corem, N.; Klochendler, A.; et al. β-Cell DNA damage response promotes islet inflammation in type 1 diabetes. Diabetes 2018, 67, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, A.; Suarez-Pinzon, W.L. Role of cytokines in the pathogenesis of autoimmune diabetes mellitus. Rev. Endocr. Metab. Disord. 2003, 4, 291–299. [Google Scholar] [CrossRef]
- Villate, O.; Turatsinze, J.V.; Mascali, L.G.; Grieco, F.A.; Nogueira, T.C.; Cunha, D.A.; Nardelli, T.R.; Sammeth, M.; Salunkhe, V.A.; Esguerra, J.L.; et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucl. Acid. Res. 2014, 42, 11818–11830. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jorgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jorgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II antigen processing and presentation pathway components demonstrated by transcriptome and protein analyses of islet beta-cells from donors with type 1 diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Souza, K.L.; Gurgul-Convey, E.; Elsner, M.; Lenzen, S. Interaction between pro-inflammatory and anti-inflammatory cytokines in insulin-producing cells. J. Endocr. 2008, 197, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Gurgul-Convey, E.; Lenzen, S. Protection against cytokine toxicity through endoplasmic reticulum and mitochondrial stress prevention by prostacyclin synthase overexpression in insulin-producing cells. J. Biol. Chem. 2010, 285, 11121–11128. [Google Scholar] [CrossRef] [Green Version]
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 2011, 89, 785–798. [Google Scholar] [CrossRef]
- Hanzelka, K.; Skalniak, L.; Jura, J.; Lenzen, S.; Gurgul-Convey, E. Effects of the novel mitochondrial protein mimitin in insulin-secreting cells. Biochem. J. 2012, 445, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Gurgul-Convey, E.; Mehmeti, I.; Plötz, T.; Jörns, A.; Lenzen, S. Sensitivity profile of the human EndoC-betaH1 beta cell line to proinflammatory cytokines. Diabetologia 2016, 59, 2125–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyka, K.; Jörns, A.; Turatsinze, J.V.; Eizirik, D.L.; Lenzen, S.; Gurgul-Convey, E. MCPIP1 regulates the sensitivity of pancreatic beta-cells to cytokine toxicity. Cell Death Dis. 2019, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, E.; Lortz, S.; Tiedge, M.; Jörns, A.; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36 Pt 3, 343–347. [Google Scholar] [CrossRef]
- Ghiasi, S.M.; Krogh, N.; Tyrberg, B.; Mandrup-Poulsen, T. The no-go and nonsense-mediated RNA decay pathways are regulated by inflammatory cytokines in insulin-producing cells and human islets and determine beta-cell insulin biosynthesis and survival. Diabetes 2018, 67, 2019–2037. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Cnop, M. ER stress in pancreatic beta cells: The thin red line between adaptation and failure. Sci. Signal. 2010, 3, pe7. [Google Scholar] [CrossRef]
- Robertson, R.P. Eicosanoids as pluripotential modulators of pancreatic islet function. Diabetes 1988, 37, 367–370. [Google Scholar] [CrossRef]
- Chambers, K.T.; Weber, S.M.; Corbett, J.A. PGJ2-stimulated beta-cell apoptosis is associated with prolonged UPR activation. Am. J. Physiol. Endocr. Metab. 2007, 292, E1052–E1061. [Google Scholar] [CrossRef]
- Heitmeier, M.R.; Kelly, C.B.; Ensor, N.J.; Gibson, K.A.; Mullis, K.G.; Corbett, J.A.; Maziasz, T.J. Role of cyclooxygenase-2 in cytokine-induced beta-cell dysfunction and damage by isolated rat and human islets. J. Biol. Chem. 2004, 279, 53145–53151. [Google Scholar] [CrossRef] [Green Version]
- Ghiasi, S.M.; Dahllof, M.S.; Osmai, Y.; Osmai, M.; Jakobsen, K.K.; Aivazidis, A.; Tyrberg, B.; Perruzza, L.; Prause, M.C.B.; Christensen, D.P.; et al. Regulation of the beta-cell inflammasome and contribution to stress-induced cellular dysfunction and apoptosis. Mol. Cell. Endocr. 2018, 478, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Sandler, S.; Andersson, A.; Hellerström, C. Inhibitory effects of interleukin 1 on insulin secretion; insulin biosynthesis; and oxidative metabolism of isolated rat pancreatic islets. Endocrinology 1987, 121, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Southern, C.; Schulster, D.; Green, I.C. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an L-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 1990, 276, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, K.; Carreras-Sureda, A.; Garcia-Lopez, R.; Rubio-Moscardo, F.; Casas, J.; Fabrias, G.; Vicente, R. Coordinated regulation of the orosomucoid-like gene family expression controls de novo ceramide synthesis in mammalian cells. J. Biol. Chem. 2015, 290, 2822–2830. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2; sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Sonoda, H.; Yu, H.; Kajimoto, T.; Goparaju, S.K.; Jahangeer, S.; Okada, T.; Nakamura, S. Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J. Biol. Chem. 2007, 282, 27493–27502. [Google Scholar] [CrossRef] [Green Version]
- Saba, J.D.; Nara, F.; Bielawska, A.; Garrett, S.; Hannun, Y.A. The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J. Biol. Chem. 1997, 272, 26087–26090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D. The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell. Signal. 2011, 23, 1144–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyaya, P.; Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D.; Hecht, S.S. The sphingolipid degradation product trans-2-hexadecenal forms adducts with DNA. Biochem. Biophys. Res. Commun. 2012, 424, 18–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, K.; Ohkuni, A.; Kitamura, T.; Abe, K.; Naganuma, T.; Ohno, Y.; Zoeller, R.A.; Kihara, A. The Sjogren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell 2012, 46, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trend Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase; a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzyme Regul. 2010, 50, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Kumagai, K.; Tomishige, N.; Kawano, M. CERT and intracellular trafficking of ceramide. BBA 2007, 1771, 644–653. [Google Scholar] [CrossRef]
- Colley, K.J.; Varki, A.; Kinoshita, T. Cellular Organization of Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 41–49. [Google Scholar]
- Gomez-Munoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordonez, M. Control of inflammatory responses by ceramide; sphingosine 1-phosphate and ceramide 1-phosphate. Progr. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef]
- Shinghal, R.; Scheller, R.H.; Bajjalieh, S.M. Ceramide 1-phosphate phosphatase activity in brain. J. Neurochem. 1993, 61, 2279–2285. [Google Scholar] [CrossRef]
- Boudker, O.; Futerman, A.H. Detection and characterization of ceramide-1-phosphate phosphatase activity in rat liver plasma membrane. J. Biol. Chem. 1993, 268, 22150–22155. [Google Scholar]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugio, L.B.; Coeli-Lacchini, F.B.; Leopoldino, A.M. Sphingolipids and mitochondrial dynamic. Cells 2020, 9, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Corbacho, M.J.; Salama, M.F.; Canals, D.; Senkal, C.E.; Obeid, L.M. Sphingolipids in mitochondria. Biochim. Biophys. Acta Mol. Cell Biol. Lipid 2017, 1862, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. BBA 2010, 1797, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Novgorodov, S.A.; Riley, C.L.; Keffler, J.A.; Yu, J.; Kindy, M.S.; Macklin, W.B.; Lombard, D.B.; Gudz, T.I. SIRT3 deacetylates ceramide synthases: Implications for mitochondrial dysfunction and brain injury. J. Biol. Chem. 2016, 291, 1957–1973. [Google Scholar] [CrossRef] [Green Version]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Müller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Oleinik, N.; Kim, J.; Roth, B.M.; Selvam, S.P.; Gooz, M.; Johnson, R.H.; Lemasters, J.J.; Ogretmen, B. Mitochondrial protein import is regulated by p17/PERMIT to mediate lipid metabolism and cellular stress. Sci. Adv. 2019, 5, eaax1978. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, J.M.; Ma, Y.; Coolen, C.; Pham, D.; Jones, C.; Lopez, A.F.; Pitson, S.M. Sphingosine and FTY720 directly bind pro-survival 14-3-3 proteins to regulate their function. Cell. Signal. 2010, 22, 1291–1299. [Google Scholar] [CrossRef]
- Habrukowich, C.; Han, D.K.; Le, A.; Rezaul, K.; Pan, W.; Ghosh, M.; Li, Z.; Dodge-Kafka, K.; Jiang, X.; Bittman, R.; et al. Sphingosine interaction with acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) regulates PP2A activity and cyclooxygenase (COX)-2 expression in human endothelial cells. J. Biol. Chem. 2010, 285, 26825–26831. [Google Scholar] [CrossRef] [Green Version]
- Keul, P.; Polzin, A.; Kaiser, K.; Gräler, M.; Dannenberg, L.; Daum, G.; Heusch, G.; Levkau, B. Potent anti-inflammatory properties of HDL in vascular smooth muscle cells mediated by HDL-S1P and their impairment in coronary artery disease due to lower HDL-S1P: A new aspect of HDL dysfunction and its therapy. FASEB J. 2019, 33, 1482–1495. [Google Scholar] [CrossRef]
- Levkau, B. Cardiovascular effects of sphingosine-1-phosphate (S1P). Handb. Exp. Pharmacol. 2013, 147–170. [Google Scholar] [CrossRef]
- Fyrst, H.; Saba, J.D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate: The Swiss army knife of sphingolipid signaling. J. Lipid Res. 2009, 50, S272–S276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Degagne, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degagne, E.; Saba, J.D. S1pping fire: Sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clin. Exp. Gastroenterol. 2014, 7, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, W.I.; Saba, J.D. S1P metabolism in cancer and other pathological conditions. Biochimie 2010, 92, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Hans, M.; Hartmann, D.; Swandulla, D.; van Echten-Deckert, G. Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Diff. 2011, 18, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Hagen, N.; Van Veldhoven, P.P.; Proia, R.L.; Park, H.; Merrill, A.H., Jr.; van Echten-Deckert, G. Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 2009, 284, 11346–11353. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Holbling, B.V.; Schumak, B.; Hubner, M.P.; Gräler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef]
- Karunakaran, I.; van Echten-Deckert, G. Sphingosine 1-phosphate–A double edged sword in the brain. BBA Biomembr. 2017, 9 Pt B, 1573–1582. [Google Scholar] [CrossRef]
- Mitroi, D.N.; Deutschmann, A.U.; Raucamp, M.; Karunakaran, I.; Glebov, K.; Hans, M.; Walter, J.; Saba, J.; Gräler, M.; Ehninger, D.; et al. Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci. Rep. 2016, 6, 37064. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, D.N.; Karunakaran, I.; Gräler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 2017, 13, 885–899. [Google Scholar] [CrossRef] [PubMed]
- van Echten-Deckert, G.; Alam, S. Sphingolipid metabolism—An ambiguous regulator of autophagy in the brain. Biol. Chem. 2018, 399, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettus, B.J.; Kitatani, K.; Chalfant, C.E.; Taha, T.A.; Kawamori, T.; Bielawski, J.; Obeid, L.M.; Hannun, Y.A. The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol. Pharmacol. 2005, 68, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H., Jr.; Futerman, A.H. Characterization of ceramide synthase 2, tissue distribution; substrate specificity; and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [Green Version]
- Etemadi, N.; Chopin, M.; Anderton, H.; Tanzer, M.C.; Rickard, J.A.; Abeysekera, W.; Hall, C.; Spall, S.K.; Wang, B.; Xiong, Y.; et al. TRAF2 regulates TNF and NF-kappaB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. eLife 2015, 4, e10592. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Milstien, S.; Grant, S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 2012, 31, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Ordonez, M.; Trueba, M.; Gomez-Munoz, A. Regulation of cell migration and inflammation by ceramide 1-phosphate. BBA 2016, 1861, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschard, K.; Blomqvist, M.; Osterbye, T.; Fredman, P. Involvement of sulfatide in beta cells and type 1 and type 2 diabetes. Diabetologia 2005, 48, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Verlaan, D.J.; Berlivet, S.; Hunninghake, G.M.; Madore, A.M.; Lariviere, M.; Moussette, S.; Grundberg, E.; Kwan, T.; Ouimet, M.; Ge, B.; et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am. J. Hum. Genet. 2009, 85, 377–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, S.; Kono, M.; Lee, Y.T.; Byrnes, C.; Li, C.; Tuymetova, G.; Proia, R.L. A genome-wide CRISPR/Cas9 screen reveals that the aryl hydrocarbon receptor stimulates sphingolipid levels. J. Biol. Chem. 2020, 295, 4341–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miani, M.; Le Naour, J.; Waeckel-Enee, E.; Verma, S.C.; Straube, M.; Emond, P.; Ryffel, B.; van Endert, P.; Sokol, H.; Diana, J. Gut microbiota-stimulated innate lymphoid cells support beta-defensin 14 expression in pancreatic endocrine cells; preventing autoimmune diabetes. Cell Metab. 2018, 28, 557–572.e6. [Google Scholar] [CrossRef] [Green Version]
- Welsh, N. Interleukin-1 beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J. Biol. Chem. 1996, 271, 8307–8312. [Google Scholar] [CrossRef] [Green Version]
- Kwon, G.; Bohrer, A.; Han, X.; Corbett, J.A.; Ma, Z.; Gross, R.W.; McDaniel, M.L.; Turk, J. Characterization of the sphingomyelin content of isolated pancreatic islets. Evaluation of the role of sphingomyelin hydrolysis in the action of interleukin-1 to induce islet overproduction of nitric oxide. BBA 1996, 1300, 63–72. [Google Scholar] [CrossRef]
- Ishizuka, N.; Yagui, K.; Tokuyama, Y.; Yamada, K.; Suzuki, Y.; Miyazaki, J.; Hashimoto, N.; Makino, H.; Saito, Y.; Kanatsuka, A. Tumor necrosis factor alpha signaling pathway and apoptosis in pancreatic beta cells. Metab. Clin. Exp. 1999, 48, 1485–1492. [Google Scholar] [CrossRef]
- Major, C.D.; Gao, Z.Y.; Wolf, B.A. Activation of the sphingomyelinase/ceramide signal transduction pathway in insulin-secreting beta-cells: Role in cytokine-induced beta-cell death. Diabetes 1999, 48, 1372–1380. [Google Scholar] [CrossRef]
- Lei, X.; Bone, R.N.; Ali, T.; Zhang, S.; Bohrer, A.; Tse, H.M.; Bidasee, K.R.; Ramanadham, S. Evidence of contribution of iPLA2beta-mediated events during islet beta-cell apoptosis due to proinflammatory cytokines suggests a role for iPLA2beta in T1D development. Endocrinology 2014, 155, 3352–3364. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bone, R.N.; Ali, T.; Wohltmann, M.; Gai, Y.; Goodwin, K.J.; Bohrer, A.E.; Turk, J.; Ramanadham, S. Genetic modulation of islet beta-cell iPLA(2)beta expression provides evidence for its impact on beta-cell apoptosis and autophagy. Islets 2013, 5, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell 2019, 177, 1536–1552.e23. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trend Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Zhu, Q.; Shan, X.; Miao, H.; Lu, Y.; Xu, J.; You, N.; Liu, C.; Liao, D.F.; Jin, J. Acute activation of acid ceramidase affects cytokine-induced cytotoxicity in rat islet beta-cells. FEBS Lett. 2009, 583, 2136–2141. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Jin, J.F.; Shan, X.H.; Liu, C.P.; Mao, X.D.; Xu, K.F.; Liu, C. Chronic activation of neutral ceramidase protects beta-cells against cytokine-induced apoptosis. Acta Pharmacol. Sin. 2008, 29, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Laychock, S.G.; Tian, Y.; Sessanna, S.M. Endothelial differentiation gene receptors in pancreatic islets and INS-1 cells. Diabetes 2003, 52, 1986–1993. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Kon, J.; Tomura, H.; Osada, M.; Murata, N.; Kuwabara, A.; Watanabe, T.; Ohta, H.; Ui, M.; Okajima, F. Activation of phospholipase C-Ca2+ system by sphingosine 1-phosphate in CHO cells transfected with Edg-3; a putative lipid receptor. FEBS Lett. 1999, 443, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Ancellin, N.; Hla, T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1; EDG-3; and EDG-5. J. Biol. Chem. 1999, 274, 18997–19002. [Google Scholar] [CrossRef] [Green Version]
- Gonda, K.; Okamoto, H.; Takuwa, N.; Yatomi, Y.; Okazaki, H.; Sakurai, T.; Kimura, S.; Sillard, R.; Harii, K.; Takuwa, Y. The novel sphingosine 1-phosphate receptor AGR16 is coupled via pertussis toxin-sensitive and -insensitive G-proteins to multiple signalling pathways. Biochem. J. 1999, 337 Pt 1, 67–75. [Google Scholar] [CrossRef]
- Toman, R.E.; Spiegel, S. Lysophospholipid receptors in the nervous system. Neurochem. Res. 2002, 27, 619–627. [Google Scholar] [CrossRef]
- Mastrandrea, L.D.; Sessanna, S.M.; Laychock, S.G. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: Response to cytokines. Diabetes 2005, 54, 1429–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laychock, S.G.; Sessanna, S.M.; Lin, M.H.; Mastrandrea, L.D. Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 2006, 147, 4705–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, D.; Nikodinovic Glumac, J.; Asselbergh, B.; Ermanoska, B.; Blocquel, D.; Steiner, R.; Estrada-Cuzcano, A.; Peeters, K.; Ooms, T.; De Vriendt, E.; et al. Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy. Neurology 2017, 88, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, R.; Hadjidemetriou, I.; Maharaj, A.; Meimaridou, E.; Buonocore, F.; Saleem, M.; Hurcombe, J.; Bierzynska, A.; Barbagelata, E.; Bergada, I.; et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 942–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Japtok, L.; Schmitz, E.I.; Fayyaz, S.; Kramer, S.; Hsu, L.J.; Kleuser, B. Sphingosine 1-phosphate counteracts insulin signaling in pancreatic beta-cells via the sphingosine 1-phosphate receptor subtype 2. FASEB J. 2015, 29, 3357–3369. [Google Scholar] [CrossRef] [Green Version]
- Cantrell Stanford, J.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Ozcan, S. Sphingosine 1-phosphate (S1P) regulates glucose-stimulated insulin secretion in pancreatic beta cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Galadari, A.; Thayyullathil, F. Role of ceramide in diabetes mellitus: Evidence and mechanisms. Lipid Health Dis. 2013, 12, 98. [Google Scholar] [CrossRef] [Green Version]
- Kavishwar, A.; Moore, A. Sphingomyelin patches on pancreatic beta-cells are indicative of insulin secretory capacity. J. Histochem. Cytochem. 2013, 61, 910–919. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöholm, A. Ceramide inhibits pancreatic beta-cell insulin production and mitogenesis and mimics the actions of interleukin-1 beta. FEBS Lett. 1995, 367, 283–286. [Google Scholar] [CrossRef] [Green Version]
- Hasan, N.M.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; Druckenbrod, N.R.; Laychock, S.G.; Mastrandrea, L.D.; MacDonald, M.J. Sphingosine kinase 1 knockdown reduces insulin synthesis and secretion in a rat insulinoma cell line. Arch. Biochem. Biophys. 2012, 518, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Supale, S.; Thorel, F.; Merkwirth, C.; Gjinovci, A.; Herrera, P.L.; Scorrano, L.; Meda, P.; Langer, T.; Maechler, P. Loss of prohibitin induces mitochondrial damages altering beta-cell function and survival and is responsible for gradual diabetes development. Diabetes 2013, 62, 3488–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, O.; Linxweiler, J.; Dudek, J.; Blum, R.; Helms, V.; et al. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012, 31, 3282–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Sandler, S.; Welsh, N.; Juntti-Berggren, L.; Berggren, P.O. Interleukin-1 beta-induced stimulation of insulin release in mouse pancreatic islets is related to diacylglycerol production and protein kinase C activation. Mol. Cell. Endocrinol. 1995, 111, 159–165. [Google Scholar] [CrossRef]
- Perrotta, C.; Bizzozero, L.; Cazzato, D.; Morlacchi, S.; Assi, E.; Simbari, F.; Zhang, Y.; Gulbins, E.; Bassi, M.T.; Rosa, P.; et al. Syntaxin 4 is required for acid sphingomyelinase activity and apoptotic function. J. Biol. Chem. 2010, 285, 40240–40251. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Ahn, M.; Afelik, S.; Becker, T.C.; Roep, B.O.; Thurmond, D.C. Syntaxin 4 expression in pancreatic beta-cells promotes islet function and protects functional beta-cell mass. Diabetes 2018, 67, 2626–2639. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, L.A.; Störling, Z.M.; Ortis, F.; Lage, K.; Bang-Berthelsen, C.; Bergholdt, R.; Hald, J.; Brorsson, C.A.; Eizirik, D.L.; Pociot, F.; et al. Huntingtin-interacting protein 14 is a type 1 diabetes candidate protein regulating insulin secretion and beta-cell apoptosis. Proc. Natl. Acad. Sci. USA 2011, 108, E681–E688. [Google Scholar] [CrossRef] [Green Version]
- Blomqvist, M.; Osterbye, T.; Mansson, J.E.; Horn, T.; Buschard, K.; Fredman, P. Sulfatide is associated with insulin granules and located to microdomains of a cultured beta cell line. Glycoconj. J. 2002, 19, 403–413. [Google Scholar] [CrossRef]
- Osterbye, T.; Jorgensen, K.H.; Fredman, P.; Tranum-Jensen, J.; Kaas, A.; Brange, J.; Whittingham, J.L.; Buschard, K. Sulfatide promotes the folding of proinsulin; preserves insulin crystals; and mediates its monomerization. Glycobiology 2001, 11, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschard, K.; Hoy, M.; Bokvist, K.; Olsen, H.L.; Madsbad, S.; Fredman, P.; Gromada, J. Sulfatide controls insulin secretion by modulation of ATP-sensitive K(+)-channel activity and Ca(2+)-dependent exocytosis in rat pancreatic beta-cells. Diabetes 2002, 51, 2514–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomqvist, M.; Carrier, M.; Andrews, T.; Pettersson, K.; Mansson, J.E.; Rynmark, B.M.; Fredman, P.; Buschard, K. In vivo administration of the C16:0 fatty acid isoform of sulfatide increases pancreatic sulfatide and enhances glucose-stimulated insulin secretion in Zucker fatty (fa/fa) rats. Diabetes Metab. Res. Rev. 2005, 21, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Palanivel, R.; Zhao, Y.; McDonald, P.; Gruber, S.; Kowluru, A. Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: Potential mechanisms underlying ceramide-mediated metabolic dysfunction of the beta cell. Apoptosis 2005, 10, 841–850. [Google Scholar] [CrossRef]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human; rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Michaud, J.; Im, D.S.; Hla, T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J. Immunol. 2010, 184, 1475–1483. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscl. Thrombos Vascul. Biol. 2007, 27, 1312–1318. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T.; Thangada, S.; Wu, M.T.; Kontos, C.D.; Wu, D.; Wu, H.; Hla, T. PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 4312–4317. [Google Scholar] [CrossRef] [Green Version]
- Windh, R.T.; Lee, M.J.; Hla, T.; An, S.; Barr, A.J.; Manning, D.R. Differential coupling of the sphingosine 1-phosphate receptors Edg-1; Edg-3; and H218/Edg-5 to the G(i); G(q); and G(12) families of heterotrimeric G proteins. J. Biol. Chem. 1999, 274, 27351–27358. [Google Scholar] [CrossRef] [Green Version]
- Gurgul-Convey, E.; Hanzelka, K.; Lenzen, S. Mechanism of prostacyclin-induced potentiation of glucose-induced insulin secretion. Endocrinology 2012, 153, 2612–2622. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, C.; He, X.; Shang, W.; Bi, Y.; Wang, D. Long-term effect of FTY720 on lymphocyte count and islet allograft survival in mice. Microsurgery 2007, 27, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Zhang, N.; Xu, J.; Shi, Q.; Ding, Y.; Bromberg, J.S. Targeting lymphangiogenesis after islet transplantation prolongs islet allograft survival. Transplantation 2011, 92, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Arntfield, M.E.; van der Kooy, D. Beta-Cell evolution: How the pancreas borrowed from the brain: The shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. Bioessays 2011, 33, 582–587. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Delahunty, G.; Wilson, G.L.; Roberts, C.T., Jr.; Shemer, J.; Hart, C.; Lesniak, M.A.; Shiloach, J.; Roth, J. Evolutionary aspects of the endocrine and nervous systems. Recent Prog. Horm. Res. 1986, 42, 549–587. [Google Scholar] [CrossRef]
- Le Roith, D.; Shiloach, J.; Roth, J. Is there an earlier phylogenetic precursor that is common to both the nervous and endocrine systems? Peptides 1982, 3, 211–215. [Google Scholar] [CrossRef]
- Rulifson, E.J.; Kim, S.K.; Nusse, R. Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science 2002, 296, 1118–1120. [Google Scholar] [CrossRef]
- Juan-Mateu, J.; Rech, T.H.; Villate, O.; Lizarraga-Mollinedo, E.; Wendt, A.; Turatsinze, J.V.; Brondani, L.A.; Nardelli, T.R.; Nogueira, T.C.; Esguerra, J.L.; et al. Neuron-enriched RNA-binding proteins regulate pancreatic beta cell function and survival. J. Biol. Chem. 2017, 292, 3466–3480. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.; Allende, M.L.; Mizukami, H.; Cook, E.K.; Gavrilova, O.; Tuymetova, G.; Clarke, B.A.; Chen, W.; Olivera, A.; Proia, R.L. Sphingosine-1-phosphate phosphatase 2 regulates pancreatic islet beta-cell endoplasmic reticulum stress and proliferation. J. Biol. Chem. 2016, 291, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Chen, J.; Lay, A.; Don, A.; Vadas, M.; Xia, P. Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic beta-cell death in diet-induced obese mice. FASEB J. 2013, 27, 4294–4304. [Google Scholar] [CrossRef]
- Song, Z.; Wang, W.; Li, N.; Yan, S.; Rong, K.; Lan, T.; Xia, P. Sphingosine kinase 2 promotes lipotoxicity in pancreatic beta-cells and the progression of diabetes. FASEB J. 2019, 33, 3636–3646. [Google Scholar] [CrossRef]
- Kacheva, S.; Lenzen, S.; Gurgul-Convey, E. Differential effects of proinflammatory cytokines on cell death and ER stress in insulin-secreting INS1E cells and the involvement of nitric oxide. Cytokine 2011, 55, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Kang, J.; Miao, H.; Feng, Y.; Xiao, L.; Hu, Z.; Liao, D.F.; Huang, Y.; Jin, J.; He, S. Low-dose cytokine-induced neutral ceramidase secretion from INS-1 cells via exosomes and its anti-apoptotic effect. FEBS J. 2014, 281, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Sysi-Aho, M.; Ermolov, A.; Gopalacharyulu, P.V.; Tripathi, A.; Seppanen-Laakso, T.; Maukonen, J.; Mattila, I.; Ruohonen, S.T.; Vahatalo, L.; Yetukuri, L.; et al. Metabolic regulation in progression to autoimmune diabetes. PLoS Comput. Biol. 2011, 7, e1002257. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Disc. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Jörns, A.; Akin, M.; Arndt, T.; Terbish, T.; Zu Vilsendorf, A.M.; Wedekind, D.; Hedrich, H.J.; Lenzen, S. Anti-TCR therapy combined with fingolimod for reversal of diabetic hyperglycemia by beta cell regeneration in the LEW.1AR1-iddm rat model of type 1 diabetes. J. Mol. Med. 2014, 92, 743–755. [Google Scholar] [CrossRef]
- Jörns, A.; Rath, K.J.; Terbish, T.; Arndt, T.; Meyer Zu Vilsendorf, A.; Wedekind, D.; Hedrich, H.J.; Lenzen, S. Diabetes prevention by immunomodulatory FTY720 treatment in the LEW.1AR1-iddm rat despite immune cell activation. Endocrinology 2010, 151, 3555–3565. [Google Scholar] [CrossRef]
- Penaranda, C.; Tang, Q.; Ruddle, N.H.; Bluestone, J.A. Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes 2010, 59, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Lenzen, S.; Arndt, T.; Elsner, M.; Wedekind, D.; Jörns, A. Rat models of human type 1 diabetes. Meth. Mol. Biol. 2020, 2128, 69–85. [Google Scholar] [CrossRef]
- MacKinnon, A.C.; Farnworth, S.L.; Hodkinson, P.S.; Henderson, N.C.; Atkinson, K.M.; Leffler, H.; Nilsson, U.J.; Haslett, C.; Forbes, S.J.; Sethi, T. Regulation of alternative macrophage activation by galectin-3. J. Immunol. 2008, 180, 2650–2658. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, A.M.; Liu, Y.; Moore, K.W.; Mui, A.L. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: Evidence for Stat3-dependent and -independent pathways. EMBO J. 1998, 17, 1006–1018. [Google Scholar] [CrossRef]
- Kaminski, A.; Kaminski, E.R.; Morgan, N.G. Pre-incubation with interleukin-4 mediates a direct protective effect against the loss of pancreatic beta-cell viability induced by proinflammatory cytokines. Clin. Exp. Immunol. 2007, 148, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, A.; Welters, H.J.; Kaminski, E.R.; Morgan, N.G. Human and rodent pancreatic beta-cells express IL-4 receptors and IL-4 protects against beta-cell apoptosis by activation of the PI3K and JAK/STAT pathways. Biosci. Rep. 2009, 30, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.A.; Morgan, N.G. The impact of anti-inflammatory cytokines on the pancreatic beta-cell. Islets 2014, 6, e950547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Thayer, T.C.; Wen, L.; Wong, F.S. Mouse models of autoimmune diabetes: The nonobese diabetic (NOD) mouse. Meth. Mol. Biol. 2020, 2128, 87–92. [Google Scholar] [CrossRef]
- Holm, L.J.; Haupt-Jorgensen, M.; Larsen, J.; Giacobini, J.D.; Bilgin, M.; Buschard, K. L-serine supplementation lowers diabetes incidence and improves blood glucose homeostasis in NOD mice. PLoS ONE 2018, 13, e0194414. [Google Scholar] [CrossRef] [Green Version]
- Lemos, J.P.; Smaniotto, S.; Messias, C.V.; Moreira, O.C.; Cotta-de-Almeida, V.; Dardenne, M.; Savino, W.; Mendes-da-Cruz, D.A. Sphingosine-1-phosphate receptor 1 is involved in non-obese diabetic mouse thymocyte migration disorders. Int. J. Mol. Sci. 2018, 19, 1446. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Bolick, D.T.; Lukashev, D.; Lappas, C.; Sitkovsky, M.; Lynch, K.R.; Hedrick, C.C. Sphingosine-1-phosphate reduces CD4+ T-cell activation in type 1 diabetes through regulation of hypoxia-inducible factor short isoform I.1 and CD69. Diabetes 2008, 57, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Buschard, K.; Hanspers, K.; Fredman, P.; Reich, E.P. Treatment with sulfatide or its precursor; galactosylceramide; prevents diabetes in NOD mice. Autoimmunity 2001, 34, 9–17. [Google Scholar] [CrossRef]
- Sharif, S.; Delovitch, T.L. Regulation of immune responses by natural killer T cells. Arch. Immunol. Ther. Exp. 2001, 49 (Suppl. 1), S23–S31. [Google Scholar]
- Sharif, S.; Arreaza, G.A.; Zucker, P.; Mi, Q.S.; Sondhi, J.; Naidenko, O.V.; Kronenberg, M.; Koezuka, Y.; Delovitch, T.L.; Gombert, J.M.; et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat. Med. 2001, 7, 1057–1062. [Google Scholar] [CrossRef]
- Mizuno, M.; Masumura, M.; Tomi, C.; Chiba, A.; Oki, S.; Yamamura, T.; Miyake, S. Synthetic glycolipid OCH prevents insulitis and diabetes in NOD mice. J. Autoimmun. 2004, 23, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Rhost, S.; Lofbom, L.; Mansson, J.; Lehuen, A.; Blomqvist, M.; Cardell, S.L. Administration of sulfatide to ameliorate type I diabetes in non-obese diabetic mice. Scand. J. Immunol. 2014, 79, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.J.; Haupt-Jorgensen, M.; Giacobini, J.D.; Hasselby, J.P.; Bilgin, M.; Buschard, K. Fenofibrate increases very-long-chain sphingolipids and improves blood glucose homeostasis in NOD mice. Diabetologia 2019, 62, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Toda, Y.; Tamatani, T.; Watanabe, T.; Suzuki, T.; Nakao, T.; Murase, K.; Kiso, M.; Hasegawa, A.; Tadano-Aritomi, K.; et al. Sulfated glycolipids are ligands for a lymphocyte homing receptor; L-selectin (LECAM-1); Binding epitope in sulfated sugar chain. Biochem. Biophys. Res. Commun. 1993, 190, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Buschard, K.; Diamant, M.; Bovin, L.E.; Mansson, J.E.; Fredman, P.; Bendtzen, K. Sulphatide and its precursor galactosylceramide influence the production of cytokines in human mononuclear cells. APMIS 1996, 104, 938–944. [Google Scholar] [CrossRef]
- Roeske-Nielsen, A.; Dalgaard, L.T.; Mansson, J.E.; Buschard, K. The glycolipid sulfatide protects insulin-producing cells against cytokine-induced apoptosis; a possible role in diabetes. Diabetes Metab. Res. Rev. 2010, 26, 631–638. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, A.; Sinioja, T.; Lamichhane, S.; Sen, P.; Bodin, J.; Siljander, H.; Dickens, A.M.; Geng, D.; Carlsson, C.; Duberg, D.; et al. Prenatal exposure to perfluoroalkyl substances modulates neonatal serum phospholipids; increasing risk of type 1 diabetes. Environ. Int. 2020, 143, 105935. [Google Scholar] [CrossRef]
- Barra, G.; Lepore, A.; Gagliardi, M.; Somma, D.; Matarazzo, M.R.; Costabile, F.; Pasquale, G.; Mazzoni, A.; Gallo, C.; Nuzzo, G.; et al. Sphingosine Kinases promote IL-17 expression in human T lymphocytes. Sci. Rep. 2018, 8, 13233. [Google Scholar] [CrossRef]
- Grieco, F.A.; Moore, F.; Vigneron, F.; Santin, I.; Villate, O.; Marselli, L.; Rondas, D.; Korf, H.; Overbergh, L.; Dotta, F.; et al. IL-17A increases the expression of proinflammatory chemokines in human pancreatic islets. Diabetologia 2014, 57, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Zheng, F. A complex auxiliary: IL-17/Th17 signaling during type 1 diabetes progression. Mol. Immunol. 2019, 105, 16–31. [Google Scholar] [CrossRef]
- Buschard, K.; Holm, L.J.; Feldt-Rasmussen, U. Insulin independence in newly diagnosed type 1 diabetes patient following fenofibrate treatment. Case Rep. Med. 2020, 2020, 6865190. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Mallela, S.K.; Ducasa, G.M.; Yoo, T.H.; Rosenfeld-Gur, E.; Zelnik, I.D.; Molina, J.; Varona Santos, J.; Ge, M.; Sloan, A.; et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat. Commun. 2019, 10, 2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, K.; Apostolopoulou, M.; Roden, M. Insulin resistance in type 1 diabetes mellitus. Metab. Clin. Exp. 2015, 64, 1629–1639. [Google Scholar] [CrossRef]
- Mehdi, A.M.; Hamilton-Williams, E.E.; Cristino, A.; Ziegler, A.; Bonifacio, E.; Le Cao, K.A.; Harris, M.; Thomas, R. A peripheral blood transcriptomic signature predicts autoantibody development in infants at risk of type 1 diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Giannopoulou, E.Z.; Winkler, C.; Chmiel, R.; Matzke, C.; Scholz, M.; Beyerlein, A.; Achenbach, P.; Bonifacio, E.; Ziegler, A.G. Islet autoantibody phenotypes and incidence in children at increased risk for type 1 diabetes. Diabetologia 2015, 58, 2317–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef] [Green Version]
- Chiarelli, F.; Giannini, C.; Primavera, M. Prediction and prevention of type 1 diabetes in children. Clin. Pediatric Endocrinol. Case Rep. Clin. Investig. 2019, 28, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Bleich, D.; Polak, M.; Chen, S.; Swiderek, K.M.; Levy-Marchal, C. Sera from children with type 1 diabetes mellitus react against a new group of antigens composed of lysophospholipids. Horm. Res. 1999, 52, 86–94. [Google Scholar] [CrossRef]
- Misasi, R.; Dionisi, S.; Farilla, L.; Carabba, B.; Lenti, L.; Di Mario, U.; Dotta, F. Gangliosides and autoimmune diabetes. Diabet Metab. Rev. 1997, 13, 163–179. [Google Scholar] [CrossRef]
- Papaccio, G. Gangliosides prevent insulitis but not islet B cell destruction in low-dose streptozocin-treated mice. Diabetes Res. Clin. Pract. 1993, 19, 9–15. [Google Scholar] [CrossRef]
- Papaccio, G.; Chieffi Baccari, G.; Mezzogiorno, V. In vivo effect of gangliosides on non-obese diabetic mice. Acta Anat. 1993, 147, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Dotta, F.; Previti, M.; Lenti, L.; Dionisi, S.; Casetta, B.; D’Erme, M.; Eisenbarth, G.S.; Di Mario, U. GM2-1 pancreatic islet ganglioside: Identification and characterization of a novel islet-specific molecule. Diabetologia 1995, 38, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Dionisi, S.; Dotta, F.; Diaz-Horta, O.; Carabba, B.; Viglietta, V.; Di Mario, U. Target antigens in autoimmune diabetes: Pancreatic gangliosides. Ann. Ist. Super. Sanita 1997, 33, 433–435. [Google Scholar] [PubMed]
- Boraska, V.; Torlak, V.; Skrabic, V.; Kacic, Z.; Jaksic, J.; Stipancic, G.; Uroic, A.S.; Markotic, A.; Zemunik, T. Glycosyltransferase B4GALNT1 and type 1 diabetes in Croatian population: Clinical investigation. Clin. Biochem. 2009, 42, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Bedia, C.; Badia, M.; Muixi, L.; Levade, T.; Tauler, R.; Sierra, A. GM2-GM3 gangliosides ratio is dependent on GRP94 through down-regulation of GM2-AP cofactor in brain metastasis cells. Sci. Rep. 2019, 9, 14241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic reticulum chaperone glucose-regulated protein 94 is essential for proinsulin handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Khilji, M.S.; Bresson, S.E.; Verstappen, D.; Pihl, C.; Andersen, P.A.K.; Agergaard, J.B.; Dahlby, T.; Bryde, T.H.; Klindt, K.; Nielsen, C.K.; et al. The inducible beta5i proteasome subunit contributes to proinsulin degradation in GRP94-deficient beta-cells and is overexpressed in type 2 diabetes pancreatic islets. Am. Physiol. Endocr. Metab. 2020, 318, E892–E900. [Google Scholar] [CrossRef]
- Nikolaeva, S.; Bayunova, L.; Sokolova, T.; Vlasova, Y.; Bachteeva, V.; Avrova, N.; Parnova, R. GM1 and GD1a gangliosides modulate toxic and inflammatory effects of E. coli lipopolysaccharide by preventing TLR4 translocation into lipid rafts. BBA 2015, 1851, 239–247. [Google Scholar] [CrossRef]
- Colli, M.L.; Nogueira, T.C.; Allagnat, F.; Cunha, D.A.; Gurzov, E.N.; Cardozo, A.K.; Roivainen, M.; Op de Beeck, A.; Eizirik, D.L. Exposure to the viral by-product dsRNA or Coxsackievirus B5 triggers pancreatic beta cell apoptosis via a Bim/Mcl-1 imbalance. PLoS Pathog. 2011, 7, e1002267. [Google Scholar] [CrossRef] [Green Version]
- Dogusan, Z.; Garcia, M.; Flamez, D.; Alexopoulou, L.; Goldman, M.; Gysemans, C.; Mathieu, C.; Libert, C.; Eizirik, D.L.; Rasschaert, J. Double-stranded RNA induces pancreatic beta-cell apoptosis by activation of the toll-like receptor 3 and interferon regulatory factor 3 pathways. Diabetes 2008, 57, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Cardozo, A.K.; Darville, M.I.; Eizirik, D.L. Double-stranded RNA cooperates with interferon-gamma and IL-1 beta to induce both chemokine expression and nuclear factor-kappa B-dependent apoptosis in pancreatic beta-cells: Potential mechanisms for viral-induced insulitis and beta-cell death in type 1 diabetes mellitus. Endocrinology 2002, 143, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Roivainen, M.; Rasilainen, S.; Ylipaasto, P.; Nissinen, R.; Ustinov, J.; Bouwens, L.; Eizirik, D.L.; Hovi, T.; Otonkoski, T. Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J. Clin. Endocr. Metab. 2000, 85, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, B.; Darville, M.I.; Cardozo, A.K.; Eizirik, D.L. Molecular regulation of monocyte chemoattractant protein-1 expression in pancreatic beta-cells. Diabetes 2003, 52, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, K.; Richardson, S.J.; Leete, P.; Morgan, N.G.; Korsgren, O.; Flodström-Tullberg, M. Induction of an antiviral state and attenuated coxsackievirus replication in type III interferon-treated primary human pancreatic islets. J. Virol. 2013, 87, 7646–7654. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Leet, E.P.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl-1. Diabetologia 2013, 56, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Morgan, N.G.; Foulis, A.K. Immunopathology of the human pancreas in type 1 diabetes. Sem. Immunopathol. 2011, 33, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.C.; Al-Shabeeb, A.; Pang, C.N.; Wilkins, M.R.; Catteau, J.; Howard, N.J.; Rawlinson, W.D.; Craig, M.E. Children with islet autoimmunity and enterovirus infection demonstrate a distinct cytokine profile. Diabetes 2012, 61, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Osterbye, T.; Funda, D.P.; Fundova, P.; Mansson, J.E.; Tlaskalova-Hogenova, H.; Buschard, K. A subset of human pancreatic beta cells express functional CD14 receptors: A signaling pathway for beta cell-related glycolipids; sulfatide and beta-galactosylceramide. Diabetes Metab. Res. Rev. 2010, 26, 656–667. [Google Scholar] [CrossRef]
- Andersson, K.; Buschard, K.; Fredman, P.; Kaas, A.; Lidström, A.M.; Madsbad, S.; Mortensen, H.; Jan-Eric, M. Patients with insulin-dependent diabetes but not those with non-insulin-dependent diabetes have anti-sulfatide antibodies as determined with a new ELISA assay. Autoimmunity 2002, 35, 463–468. [Google Scholar] [CrossRef]
- Blomqvist, M.; Kaas, A.; Mansson, J.E.; Formby, B.; Rynmark, B.M.; Buschard, K.; Fredman, P. Developmental expression of the type I diabetes related antigen sulfatide and sulfated lactosylceramide in mammalian pancreas. J. Cell. Biochem. 2003, 89, 301–310. [Google Scholar] [CrossRef]
- Kavishwar, A.; Medarova, Z.; Moore, A. Unique sphingomyelin patches are targets of a beta-cell-specific antibody. J. Lipid Res. 2011, 52, 1660–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigger, D.; Gulbins, E.; Kleuser, B.; Schumacher, F. Monitoring the sphingolipid de novo synthesis by stable-isotope labeling and liquid chromatography-mass spectrometry. Front. Cell Dev. Biol. 2019, 7, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parashuraman, S.; D’Angelo, G. Visualizing sphingolipid biosynthesis in cells. Chem. Phys. Lipid 2019, 218, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef]
- Gurgul-Convey, E.; Kaminski, M.T.; Lenzen, S. Physiological characterization of the human EndoC-betaH1 beta-cell line. Biochem. Biophys. Res. Commun. 2015, 464, 13–19. [Google Scholar] [CrossRef]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurgul-Convey, E. Sphingolipids in Type 1 Diabetes: Focus on Beta-Cells. Cells 2020, 9, 1835. https://doi.org/10.3390/cells9081835
Gurgul-Convey E. Sphingolipids in Type 1 Diabetes: Focus on Beta-Cells. Cells. 2020; 9(8):1835. https://doi.org/10.3390/cells9081835
Chicago/Turabian StyleGurgul-Convey, Ewa. 2020. "Sphingolipids in Type 1 Diabetes: Focus on Beta-Cells" Cells 9, no. 8: 1835. https://doi.org/10.3390/cells9081835
APA StyleGurgul-Convey, E. (2020). Sphingolipids in Type 1 Diabetes: Focus on Beta-Cells. Cells, 9(8), 1835. https://doi.org/10.3390/cells9081835