The Impact of ALS-Associated Genes hnRNPA1, MATR3, VCP and UBQLN2 on the Severity of TDP-43 Aggregation


Abstract
:
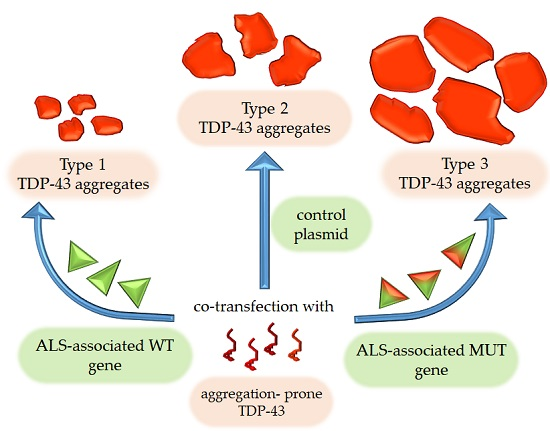{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. Western Blot
2.4. Immunocytochemistry
2.5. Imaging and Statistical Analysis
3. Results
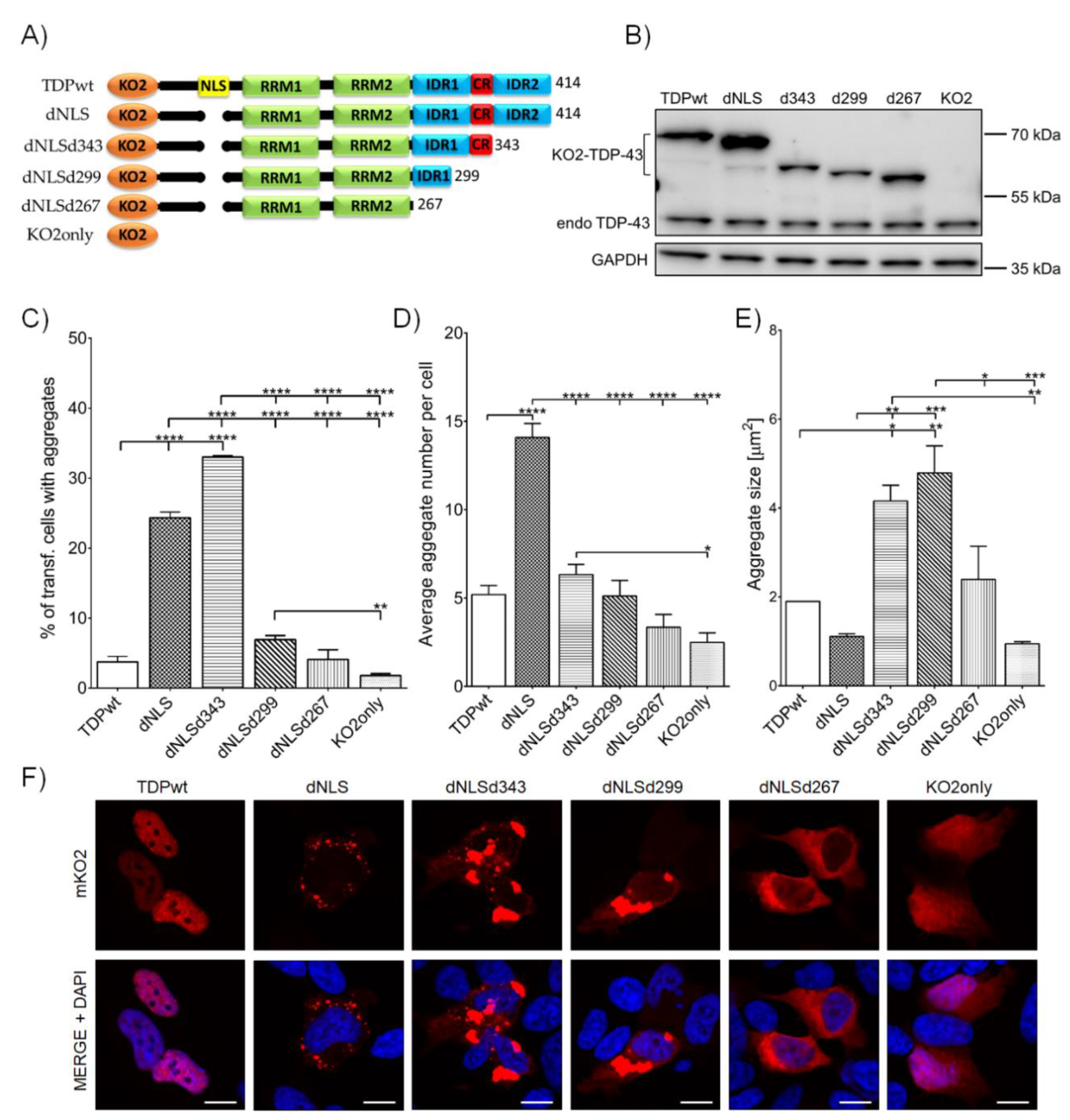
3.1. TDP-43 Aggregation Is Affected by Shortening of the C-Terminal Domain
3.2. The Mutations of ALS-Associated Genes Display Impact on TDP-43 Aggregation Behavior
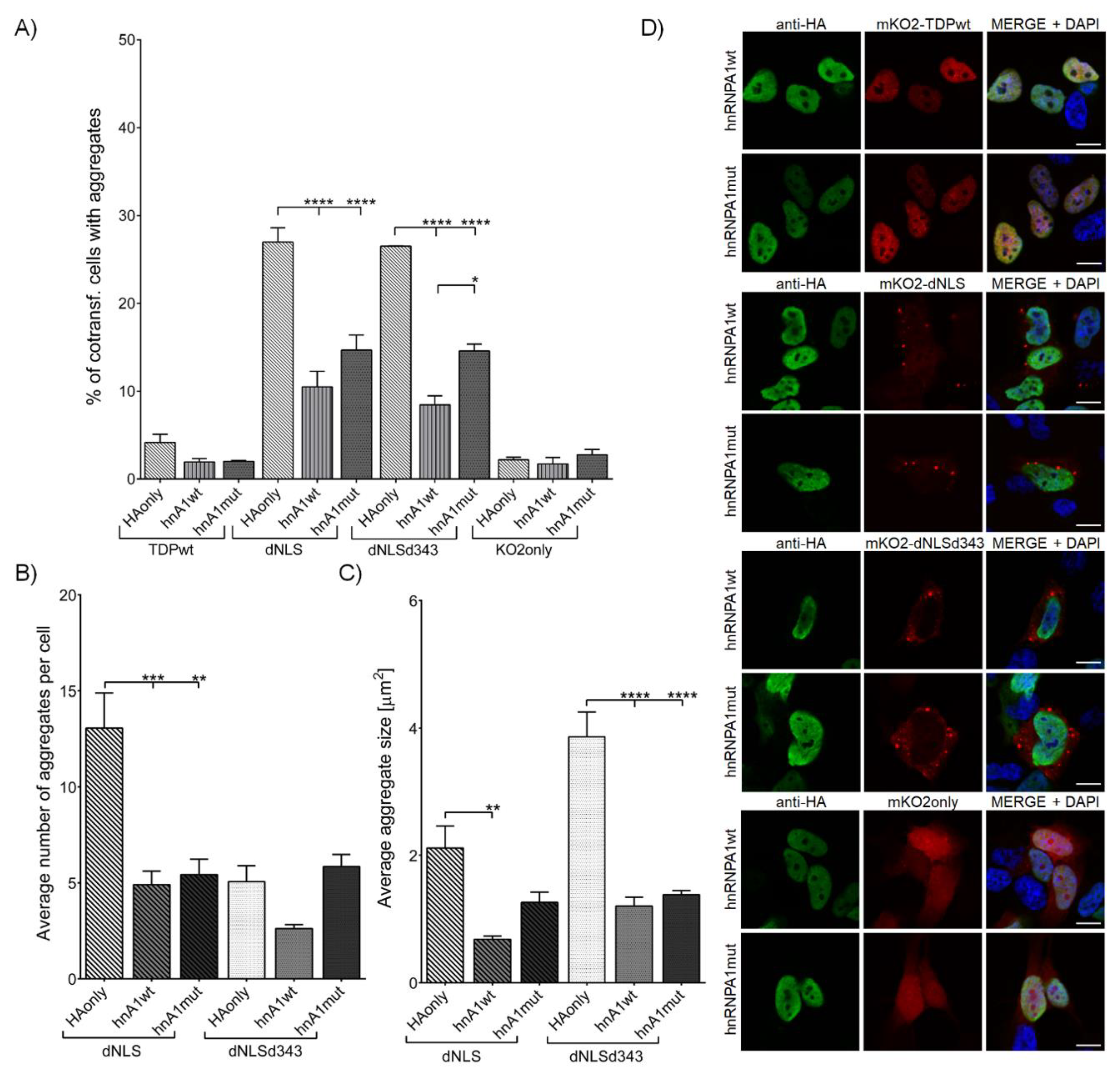
3.2.1. Overexpression of hnRNPA1 and Its D262V Mutation Inhibits TDP-43 Aggregate Formation and Their Maturation
3.2.2. MATR3 S85C Mutation Promotes TDP-43 Aggregation
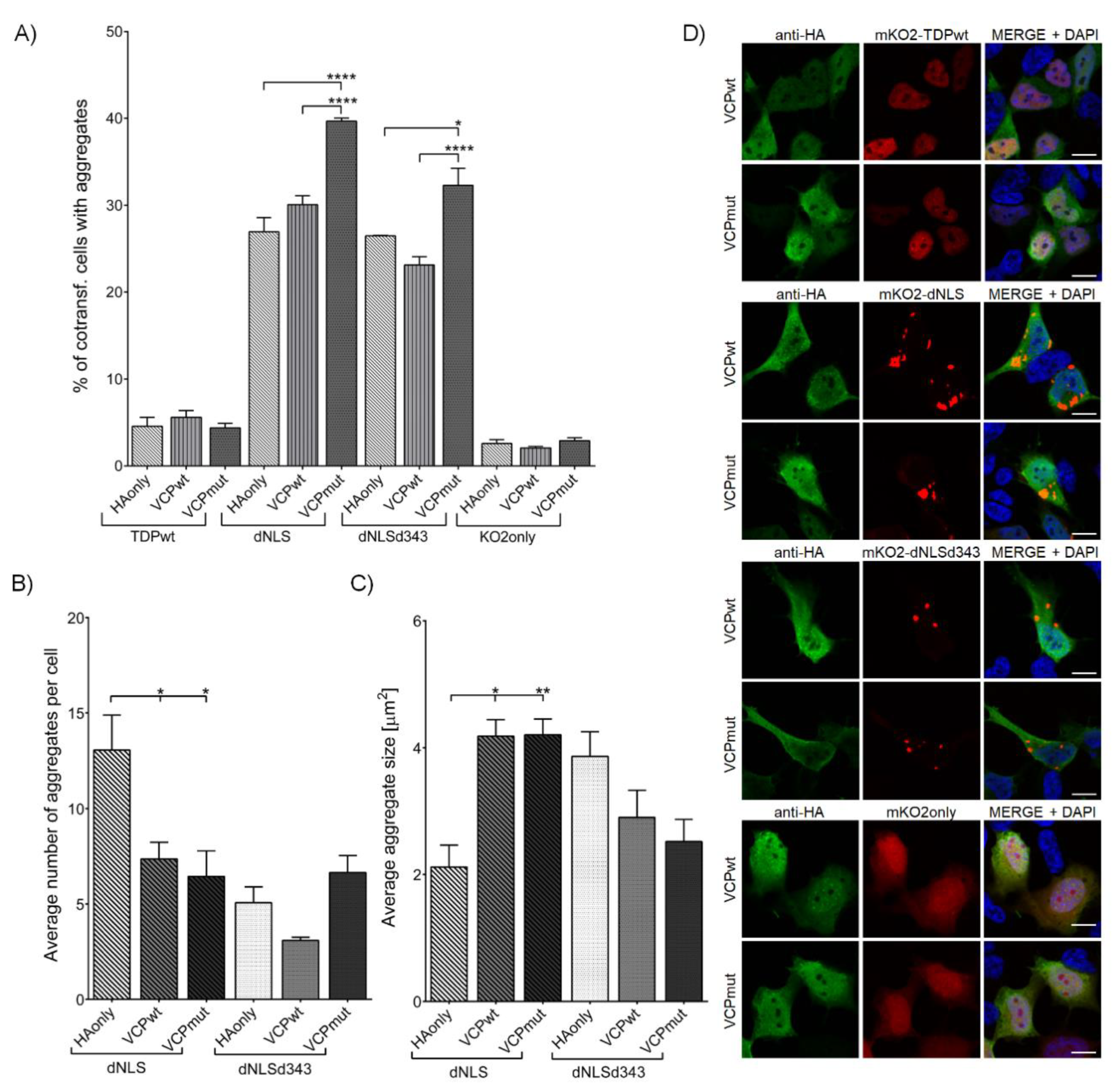
3.2.3. VCP R191Q Mutation Promotes TDP-43 dNLS but Not dNLSd343 Aggregate Maturation
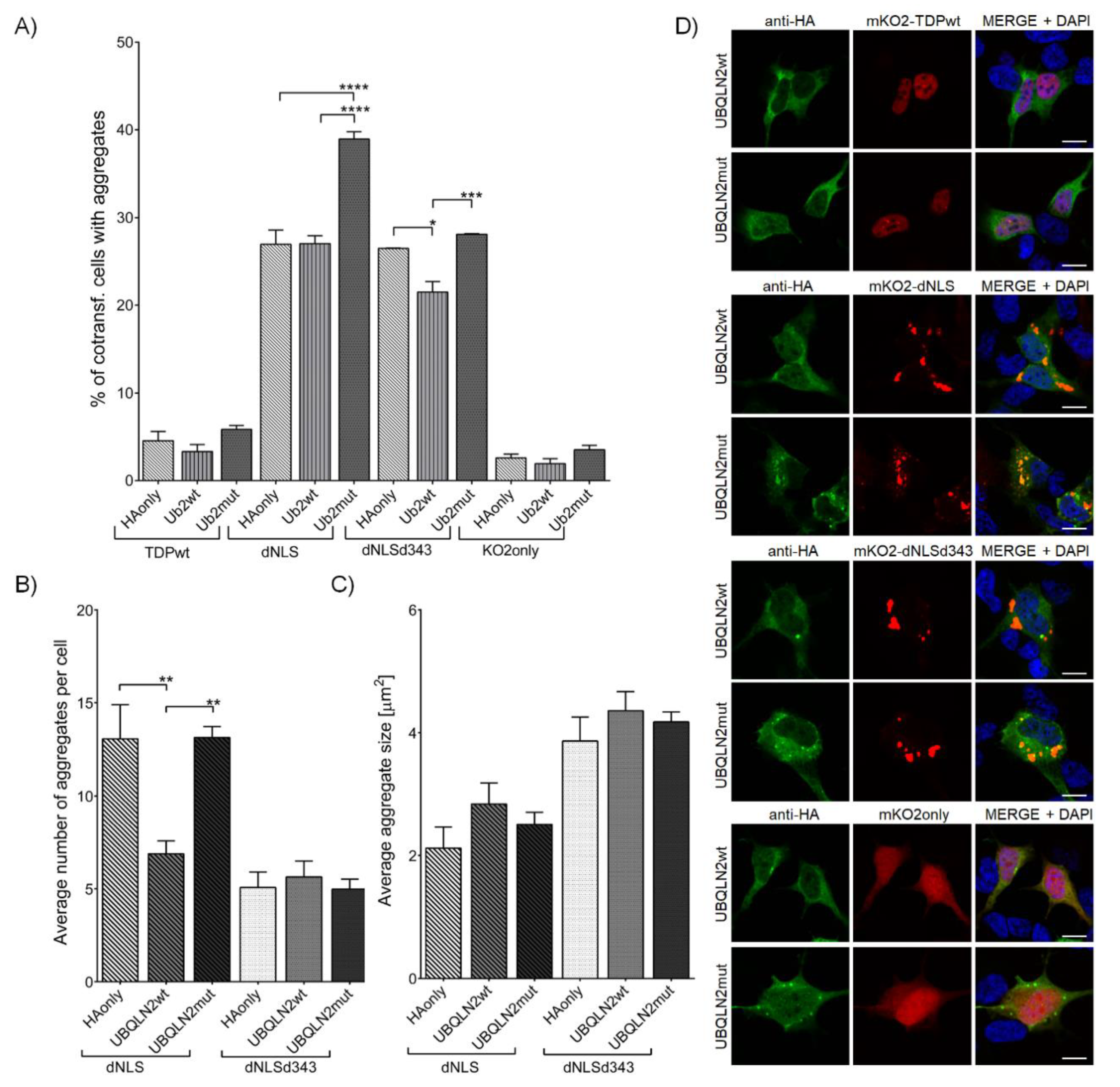
3.2.4. Wild-Type UBQLN2, but Not Its P506T Mutation Decreases Initiation of TDP-43 dNLS Aggregate Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Loughlin, F.E.; Wilce, J.A. TDP-43 and FUS-structural insights into RNA recognition and self-association. Curr. Opin. Struct. Biol. 2019, 59, 134–142. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Župunski, V.; Troakes, C.; Kathe, C.; Fratta, P.; Howell, M.; Gallo, J.-M.; Hortobágyi, T.; Shaw, C.E.; Rogelj, B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 2010, 133, 1763–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modic, M.; Grosch, M.; Rot, G.; Schirge, S.; Lepko, T.; Yamazaki, T.; Lee, F.C.Y.; Rusha, E.; Shaposhnikov, D.; Palo, M.; et al. Cross-regulation between TDP-43 and paraspeckles promotes pluripotency-differentiation transition. Mol. Cell. 2019, 74, 951–965.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Štalekar, M.; Yin, X.; Rebolj, K.; Darovic, S.; Troakes, C.; Mayr, M.; Shaw, C.E.; Rogelj, B. Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 2015, 293, 157–170. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Shum, C.; Scotter, E.L.; Abdelgany, A.; Sardone, V.; Wright, J.; Lee, Y.-B.; Chen, H.-J.; Bilican, B.; Carrasco, M.; et al. Allele-specific knockdown of ALS-associated mutant TDP-43 in neural stem cells derived from induced pluripotent stem cells. PLoS ONE 2014, 9, e91269. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.J.; et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darovic, S.; Prpar Mihevc, S.; Župunski, V.; Gunčar, G.; Štalekar, M.; Lee, Y.-B.; Shaw, C.E.; Rogelj, B. Phosphorylation of C-terminal tyrosine residue 526 in FUS impairs its nuclear import. J. Cell. Sci. 2015, 128, 4151–4159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N.; et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012, 2, 603. [Google Scholar] [CrossRef]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Mediani, L.; Alberti, S.; Carra, S. ALS and FTD: Where RNA metabolism meets protein quality control. Semin. Cell Dev. Biol. 2020, 99, 183–192. [Google Scholar] [CrossRef]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Babinchak, W.M.; Surewicz, W.K. Liquid-liquid phase separation and its mechanistic role in pathological protein aggregation. J. Mol. Biol. 2020, 432, 1910–1925. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.-K.; Surewicz, W.K. The role of liquid–liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.B.; Barreau, A.; Rohatgi, R. Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun. 2019, 10, 4890. [Google Scholar] [CrossRef] [Green Version]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-R.; Chen, T.-C.; Hsiao, C.-L.; Shi, L.; Chou, C.-Y.; Huang, J. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2018, 1866, 214–223. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, P.S.; Choi, K.-J.; Leonard, P.G.; Sizovs, A.; Moosa, M.M.; MacKenzie, K.R.; Ferreon, J.C.; Ferreon, A.C.M. The N-terminal domain of ALS-linked TDP-43 assembles without Misfolding. Angew. Chem. Int. Ed. Engl. 2017, 56, 12590–12593. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Caulfield, T.; Xu, Y.-F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Arslan, P.E.; Won, A.; Yip, C.M.; Chakrabartty, A. Binding of TDP-43 to the 3’UTR of its cognate mRNA enhances its solubility. Biochemistry 2014, 53, 5885–5894. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Lin, K.-F.; He, R.-Y.; Tu, P.-H.; Koubek, J.; Hsu, Y.-C.; Huang, J.J.-T. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS ONE 2013, 8, e64002. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [Green Version]
- French, R.L.; Grese, Z.R.; Aligireddy, H.; Dhavale, D.D.; Reeb, A.N.; Kedia, N.; Kotzbauer, P.T.; Bieschke, J.; Ayala, Y.M. Detection of TAR DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J. Biol. Chem. 2019, 294, 6696–6709. [Google Scholar] [CrossRef]
- Solomon, D.A.; Stepto, A.; Au, W.H.; Adachi, Y.; Diaper, D.C.; Hall, R.; Rekhi, A.; Boudi, A.; Tziortzouda, P.; Lee, Y.-B.; et al. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-α mediates C9orf72-related neurodegeneration. Brain 2018, 141, 2908–2924. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, T.; Hasegawa, M. Prion-like properties of assembled TDP-43. Curr. Opin. Neurobiol. 2020, 61, 23–28. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Watanabe, S.; Yamanaka, K.; Nukina, N. A Seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble Transactivation Response Element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 2011, 286, 18664–18672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M.-Y. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.A.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell. Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.-R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.-M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Kitagawa, K.; Arai, K. Phenotypic variability and its pathological basis in amyotrophic lateral sclerosis. Neuropathology 2020, 40, 40–56. [Google Scholar] [CrossRef]
- Velebit, J.; Horvat, A.; Smolič, T.; Prpar Mihevc, S.; Rogelj, B.; Zorec, R.; Vardjan, N. Astrocytes with TDP-43 inclusions exhibit reduced noradrenergic cAMP and Ca2+ signaling and dysregulated cell metabolism. Sci. Rep. 2020, 10, 6003. [Google Scholar] [CrossRef] [Green Version]
- Mihevc, S.P.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 2016, 6, 33996. [Google Scholar] [CrossRef]
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323. [Google Scholar] [CrossRef] [Green Version]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.-P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Kleijnen, M.F.; Shih, A.H.; Zhou, P.; Kumar, S.; Soccio, R.E.; Kedersha, N.L.; Gill, G.; Howley, P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 2000, 6, 409–419. [Google Scholar] [CrossRef]
- Salton, M.; Elkon, R.; Borodina, T.; Davydov, A.; Yaspo, M.-L.; Halperin, E.; Shiloh, Y. Matrin 3 Binds and Stabilizes mRNA. PLoS ONE 2011, 6, e23882. [Google Scholar] [CrossRef] [Green Version]
- Jaunmuktane, Z.; Brandner, S. Invited review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Bekenstein, U.; Soreq, H. Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: From structural insights to post-transcriptional regulatory roles. Mol. Cell. Neurosci. 2013, 56, 436–446. [Google Scholar] [CrossRef]
- Howard, J.M.; Lin, H.; Wallace, A.J.; Kim, G.; Draper, J.M.; Haeussler, M.; Katzman, S.; Toloue, M.; Liu, Y.; Sanford, J.R. HNRNPA1 promotes recognition of splice site decoys by U2AF2 in vivo. Genome Res. 2018, 28, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Huang, Y.; Seckl, M.J.; Pardo, O.E. Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kattuah, W.; Rogelj, B.; King, A.; Shaw, C.E.; Hortobágyi, T.; Troakes, C. Heterogeneous nuclear Ribonucleoprotein E2 (hnRNP E2) is a component of TDP-43 aggregates specifically in the A and C pathological subtypes of Frontotemporal lobar degeneration. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef] [Green Version]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Cruz, S.D.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and maturation of phase separated liquid droplets by RNA binding proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salton, M.; Lerenthal, Y.; Wang, S.-Y.; Chen, D.J.; Shiloh, Y. Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 2010, 9, 1568–1576. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.B.; Attig, J.; Bellora, N.; König, J.; Hallegger, M.; Kayikci, M.; Eyras, E.; Ule, J.; Smith, C.W.J. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 2015, 34, 653–668. [Google Scholar] [CrossRef]
- Attig, J.; Agostini, F.; Gooding, C.; Chakrabarti, A.M.; Singh, A.; Haberman, N.; Zagalak, J.A.; Emmett, W.; Smith, C.W.J.; Luscombe, N.M.; et al. Heteromeric RNP assembly at LINEs controls lineage-specific RNA processing. Cell 2018, 174, 1067–1081.e17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Kim, J.R.; van Bruggen, R.; Park, J. RNA-binding proteins in amyotrophic lateral sclerosis. Mol. Cells 2018, 41, 818–829. [Google Scholar] [CrossRef]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA buffers the phase separation behavior of prion-like RNA-binding proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehringer, A.; Garcia-Mansfield, K.; Singh, G.; Bakkar, N.; Pirrotte, P.; Bowser, R. ALS associated mutations in Matrin 3 alter protein-protein interactions and impede mRNA nuclear export. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Iradi, M.C.G.; Triplett, J.C.; Thomas, J.D.; Davila, R.; Crown, A.M.; Brown, H.; Lewis, J.; Swanson, M.S.; Xu, G.; Rodriguez-Lebron, E.; et al. Characterization of gene regulation and protein interaction networks for Matrin 3 encoding mutations linked to amyotrophic lateral sclerosis and myopathy. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Iradi, M.C.; Strunk, H.; Crown, A.M.; Davila, R.; Brown, H.; Rodriguez-Lebron, E.; Borchelt, D.R. N-terminal sequences in Matrin 3 mediate phase separation into droplet-like structures that recruit TDP43 variants lacking RNA binding elements. Lab. Investig. 2019, 99, 1030–1040. [Google Scholar] [CrossRef]
- Malik, A.M.; Miguez, R.A.; Li, X.; Ho, Y.-S.; Feldman, E.L.; Barmada, S.J. Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. eLife 2018, 7, e35977. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 414652a. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A mighty “Protein Extractor” of the cell: Structure and function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.-B.; Chen, H.-J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wang, Q.; Li, C.-C.H. Characterization of the aggregation-prevention activity of p97/valosin-containing protein. Biochemistry 2007, 46, 14889–14898. [Google Scholar] [CrossRef]
- Seguin, S.J.; Morelli, F.F.; Vinet, J.; Amore, D.; De Biasi, S.; Poletti, A.; Rubinsztein, D.C.; Carra, S. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 2014, 21, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Kolaitis, R.-M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Maxwell, B.A.; Joo, J.H.; Gwon, Y.; Messing, J.; Mishra, A.; Shaw, T.I.; Ward, A.L.; Quan, H.; Sakurada, S.M.; et al. ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol. Cell 2019, 74, 742–757.e8. [Google Scholar] [CrossRef]
- Dobra, I.; Pankivskyi, S.; Samsonova, A.; Pastre, D.; Hamon, L. Relation between stress granules and cytoplasmic protein aggregates linked to neurodegenerative diseases. Curr. Neurol. Neurosci. Rep. 2018, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 Aggregation in neurodegeneration: Are stress granules the key? Brain Res. 2012, 1462, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Yang, S.; Warraich, S.T.; Blair, I.P. Ubiquilin 2: A component of the ubiquitin–proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell Biol. 2014, 50, 123–126. [Google Scholar] [CrossRef]
- Alexander, E.J.; Ghanbari Niaki, A.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.P.; Kolaitis, R.-M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castañeda, C.A. Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol. Cell 2018, 69, 965–978.e6. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.P.; Martyniak, B.; Canning, A.J.; Lei, Y.; Colicino, E.G.; Cosgrove, M.S.; Hehnly, H.; Castañeda, C.A. ALS-linked mutations affect UBQLN2 Oligomerization and phase separation in a position- and amino acid-dependent manner. Structure 2019, 27, 937–951.e5. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajc Česnik, A.; Motaln, H.; Rogelj, B. The Impact of ALS-Associated Genes hnRNPA1, MATR3, VCP and UBQLN2 on the Severity of TDP-43 Aggregation. Cells 2020, 9, 1791. https://doi.org/10.3390/cells9081791
Bajc Česnik A, Motaln H, Rogelj B. The Impact of ALS-Associated Genes hnRNPA1, MATR3, VCP and UBQLN2 on the Severity of TDP-43 Aggregation. Cells. 2020; 9(8):1791. https://doi.org/10.3390/cells9081791
Chicago/Turabian StyleBajc Česnik, Ana, Helena Motaln, and Boris Rogelj. 2020. "The Impact of ALS-Associated Genes hnRNPA1, MATR3, VCP and UBQLN2 on the Severity of TDP-43 Aggregation" Cells 9, no. 8: 1791. https://doi.org/10.3390/cells9081791
APA StyleBajc Česnik, A., Motaln, H., & Rogelj, B. (2020). The Impact of ALS-Associated Genes hnRNPA1, MATR3, VCP and UBQLN2 on the Severity of TDP-43 Aggregation. Cells, 9(8), 1791. https://doi.org/10.3390/cells9081791