Endocytosis: A Turnover Mechanism Controlling Ion Channel Function

 , ,
, , 
Abstract
:1. Introduction
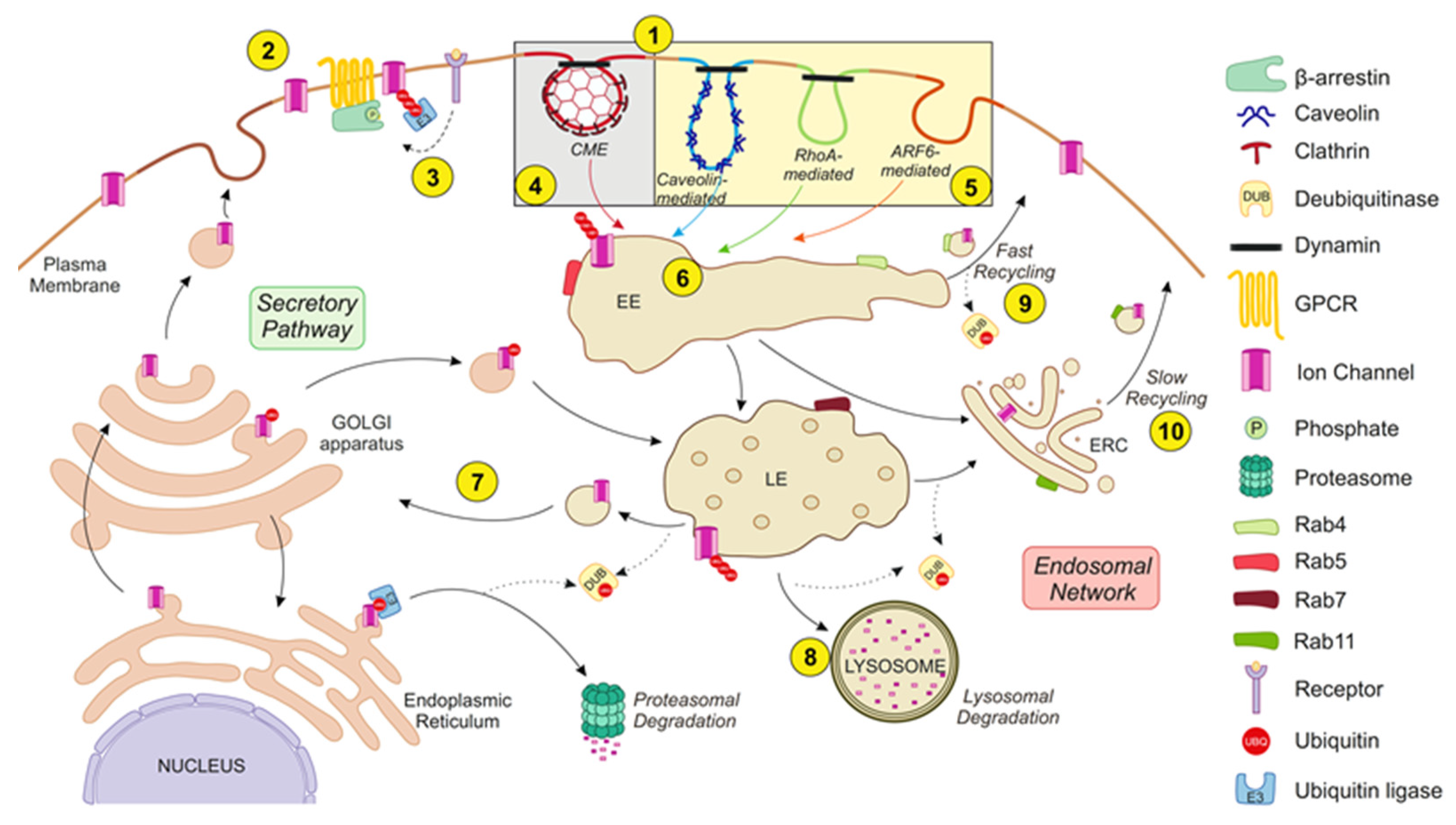
2. Endocytic Pathways
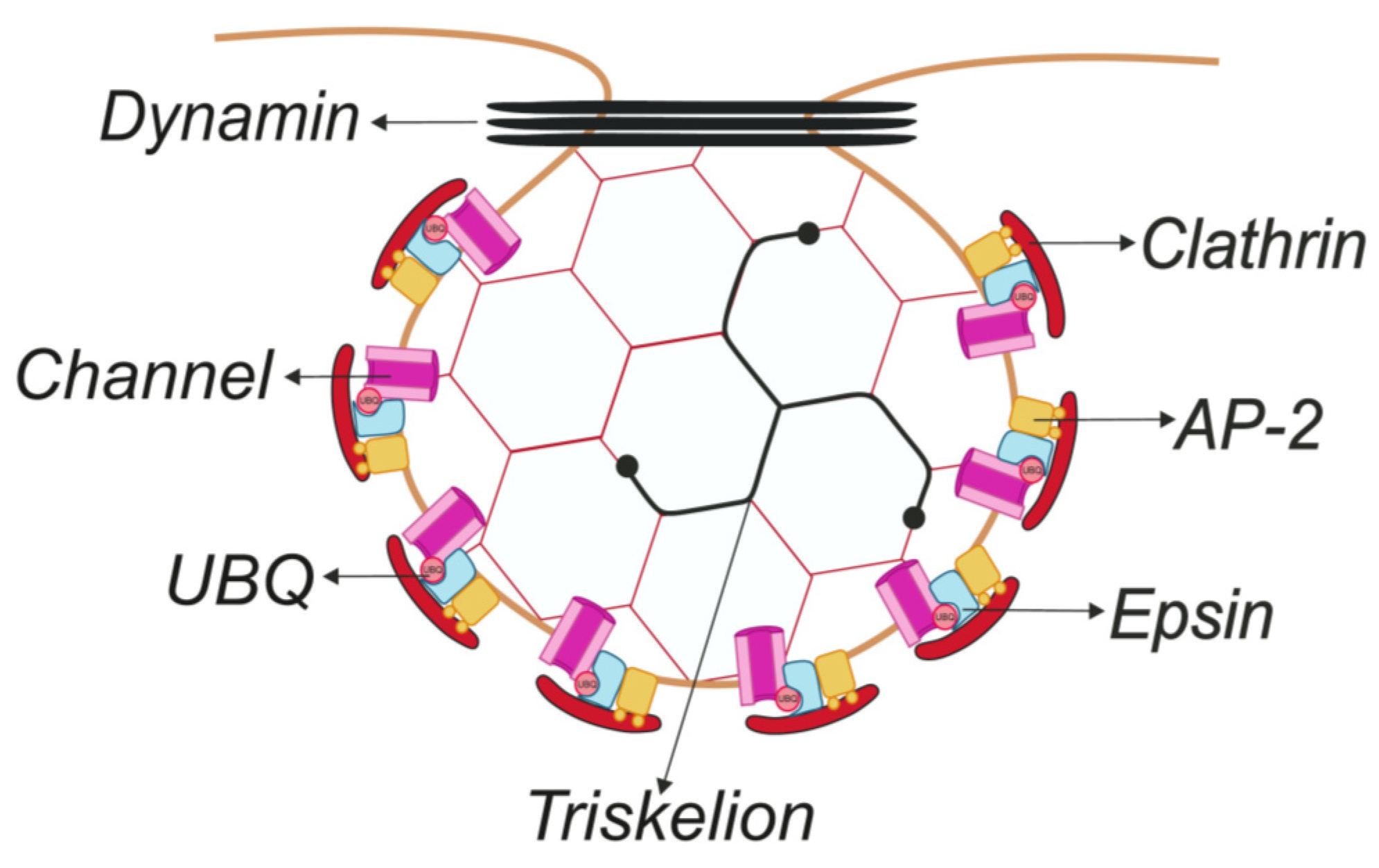
2.1. Clathrin-Dependent Endocytosis
2.1.1. Linear Sequences
2.1.2. Ubiquitination
2.2. Clathrin-Independent Endocytosis
2.2.1. Mechanisms Dependent on Caveolin
2.2.2. The RhoA-Dependent Mechanism
2.2.3. The ARF6-Dependent Pathway
3. Stimulus-Induced Endocytosis of Ion Channels
3.1. Receptor-Mediated Internalization
3.2. Drug-Induced Endocytosis
3.3. Low K+-Induced Endocytosis
4. The Components of Ion Channel Endocytosis
4.1. Ubiquitin Ligases and Deubiquinating Enzymes
4.2. β-Arrestins
4.3. Rab Proteins
4.3.1. Rab4
4.3.2. Rab5
4.3.3. Rab7
4.3.4. Rab11
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hille, B. Ion. Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, INC: Sunderland, MA, USA, 2001. [Google Scholar]
- Zheng, J.; Trudeau, M.C. Handbook of Ion Channels; Taylor & Francis Goup, LLC: Boca Raton, FL, USA, 2015. [Google Scholar]
- Lang, F.; Foller, M.; Lang, K.; Lang, P.; Ritter, M.; Vereninov, A.; Szabo, I.; Huber, S.M.; Gulbins, E. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol. 2007, 428, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Panyi, G.; Varga, Z.; Gáspár, R. Ion channels and lymphocyte activation. Immunol. Lett. 2004, 92, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Norota, I.; Obara, Y. Endocytic regulation of voltage-dependent potassium channels in the heart. J. Pharmacol. Sci. 2012, 120, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capera, J.; Serrano-Novillo, C.; Navarro-Perez, M.; Cassinelli, S.; Felipe, A. The Potassium Channel Odyssey: Mechanisms of Traffic and Membrane Arrangement. Int. J. Mol. Sci. 2019, 20, 734. [Google Scholar] [CrossRef] [Green Version]
- Mankouri, J.; Taneja, T.K.; Smith, A.J.; Ponnambalam, S.; Sivaprasadarao, A. Kir6.2 mutations causing neonatal diabetes prevent endocytosis of ATP-sensitive potassium channels. Embo J. 2006, 25, 4142–4151. [Google Scholar] [CrossRef] [PubMed]
- Shimkets, R.A.; Warnock, D.G.; Bositis, C.M.; Nelson-Williams, C.; Hansson, J.H.; Schambelan, M.; Gill, J.R., Jr.; Ulick, S.; Milora, R.V.; Findling, J.W.; et al. Liddle’s syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 1994, 79, 407–414. [Google Scholar] [CrossRef]
- Dennis, A.; Wang, L.; Wan, X.; Ficker, E. hERG channel trafficking: Novel targets in drug-induced long QT syndrome. Biochem. Soc. Trans. 2007, 35, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Naslavsky, N.; Caplan, S. The enigmatic endosome—Sorting the ins and outs of endocytic trafficking. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Shimkets, R.A.; Lifton, R.P.; Canessa, C.M. The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J. Biol. Chem. 1997, 272, 25537–25541. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Mármol, R.; Styrczewska, K.; Pérez-Verdaguer, M.; Vallejo-Gracia, A.; Comes, N.; Sorkin, A.; Felipe, A. Ubiquitination mediates Kv1.3 endocytosis as a mechanism for protein kinase C-dependent modulation. Sci. Rep. 2017, 7, 42395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurakami, K.; Norota, I.; Nasu, F.; Ohshima, S.; Nagasawa, Y.; Konno, Y.; Obara, Y.; Ishii, K. KCNQ1 is internalized by activation of alpha1 adrenergic receptors. Biochem. Pharm. 2019, 169, 113628. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, N.A.; Cohn, J.A.; Venglarik, C.J.; Bridges, R.J. Biochemical and biophysical identification of cystic fibrosis transmembrane conductance regulator chloride channels as components of endocytic clathrin-coated vesicles. J. Biol. Chem. 1994, 269, 8296–8302. [Google Scholar] [PubMed]
- Conrad, R.; Stolting, G.; Hendriks, J.; Ruello, G.; Kortzak, D.; Jordan, N.; Gensch, T.; Hidalgo, P. Rapid Turnover of the Cardiac L-Type CaV1.2 Channel by Endocytic Recycling Regulates Its Cell Surface Availability. iScience 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, A.K.; Jacobs, B.E.; Welling, P.A. AP-2-dependent Internalization of Potassium Channel Kir2.3 Is Driven by a Novel Di-hydrophobic Signal. J. Biol. Chem. 2008, 283, 5973–5984. [Google Scholar] [CrossRef] [Green Version]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar] [CrossRef] [Green Version]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Takei, K.; Haucke, V. Clathrin-mediated endocytosis: Membrane factors pull the trigger. Trends Cell Biol. 2001, 11, 385–391. [Google Scholar] [CrossRef]
- Sorkin, A.; Puthenveedu, M.A. Clathrin-Mediated Endocytosis. In Vesicle Trafficking in Cancer; Yarden, Y., Tarcic, G., Eds.; Springer: New York, NY, USA, 2013; pp. 1–31. [Google Scholar] [CrossRef]
- Sorkin, A. Cargo recognition during clathrin-mediated endocytosis: A team effort. Curr. Opin. Cell Biol. 2004, 16, 392–399. [Google Scholar] [CrossRef]
- Traub, L.M.; Bonifacino, J.S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790. [Google Scholar] [CrossRef]
- Fang, L.; Garuti, R.; Kim, B.Y.; Wade, J.B.; Welling, P.A. The ARH adaptor protein regulates endocytosis of the ROMK potassium secretory channel in mouse kidney. J. Clin. Investig. 2009, 119, 3278–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, H.; Harada, K.; Nakamura, J.; Inoue, M. Nerve growth factor-induced endocytosis of TWIK-related acid-sensitive K(+) 1 channels in adrenal medullary cells and PC12 cells. Pflug. Arch. 2013, 465, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Woelk, T.; Sigismund, S.; Penengo, L.; Polo, S. The ubiquitination code: A signalling problem. Cell Div. 2007, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millard, S.M.; Wood, S.A. Riding the DUBway: Regulation of protein trafficking by deubiquitylating enzymes. J. Cell Biol. 2006, 173, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foot, N.; Henshall, T.; Kumar, S. Ubiquitination and the Regulation of Membrane Proteins. Physiol Rev. 2017, 97, 253–281. [Google Scholar] [CrossRef] [Green Version]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef]
- Hicke, L.; Dunn, R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 2003, 19, 141–172. [Google Scholar] [CrossRef]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef]
- Owen, D.J.; Collins, B.M.; Evans, P.R. Adaptors for clathrin coats: Structure and function. Annu. Rev. Cell Dev. Biol. 2004, 20, 153–191. [Google Scholar] [CrossRef]
- Garty, H.; Palmer, L.G. Epithelial sodium channels: Function, structure, and regulation. Physiol. Rev. 1997, 77, 359–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Traub, L.M.; Weixel, K.M.; Hawryluk, M.J.; Shah, N.; Edinger, R.S.; Perry, C.J.; Kester, L.; Butterworth, M.B.; Peters, K.W.; et al. Clathrin-mediated endocytosis of the epithelial sodium channel. Role of epsin. J. Biol. Chem. 2006, 281, 14129–14135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staruschenko, A.; Pochynyuk, O.; Stockand, J.D. Regulation of epithelial Na+ channel activity by conserved serine/threonine switches within sorting signals. J. Biol. Chem. 2005, 280, 39161–39167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado-Baez, L.; Williamson, C.; Donaldson, J.G. Clathrin-independent endocytosis: A cargo-centric view. Exp. Cell Res. 2013, 319, 2759–2769. [Google Scholar] [CrossRef] [Green Version]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Jiao, J.; Garg, V.; Yang, B.; Elton, T.S.; Hu, K. Protein kinase C-epsilon induces caveolin-dependent internalization of vascular adenosine 5′-triphosphate-sensitive K+ channels. Hypertension 2008, 52, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.K.; Wu, T.; Huang, C.L. Protein kinase C inhibits caveolae-mediated endocytosis of TRPV5. Am. J. Physiol. Ren. Physiol. 2008, 294, F1212–F1221. [Google Scholar] [CrossRef]
- Van de Graaf, S.F.; Rescher, U.; Hoenderop, J.G.; Verkaart, S.; Bindels, R.J.; Gerke, V. TRPV5 is internalized via clathrin-dependent endocytosis to enter a Ca2+-controlled recycling pathway. J. Biol. Chem. 2008, 283, 4077–4086. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.Z.; Babich, V.; Ortega, B.; Quigley, R.; White, S.J.; Welling, P.A.; Huang, C.L. Evidence for endocytosis of ROMK potassium channel via clathrin-coated vesicles. Am. J. Physiol. Ren. Physiol. 2002, 283, F630–F639. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.H.; Yue, P.; Pan, C.; Sun, P.; Wang, W.H. MicroRNA 802 stimulates ROMK channels by suppressing caveolin-1. J. Am. Soc. Nephrol. 2011, 22, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirling, L.; Williams, M.R.; Morielli, A.D. Dual roles for RHOA/RHO-kinase in the regulated trafficking of a voltage-sensitive potassium channel. Mol. Biol. Cell 2009, 20, 2991–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Q.; Weide, M.; Huntsman, C.; Xu, Z.; Jan, L.Y.; Ma, D. Identification and characterization of a new class of trafficking motifs for controlling clathrin-independent internalization and recycling. J. Biol. Chem. 2007, 282, 13087–13097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Karnik, R.; Ludlow, M.J.; Abuarab, N.; Smith, A.J.; Hardy, M.E.; Elliott, D.J.; Sivaprasadarao, A. Endocytosis of HERG is clathrin-independent and involves arf6. PLoS ONE 2013, 8, e85630. [Google Scholar] [CrossRef]
- Nesti, E.; Everill, B.; Morielli, A.D. Endocytosis as a Mechanism for Tyrosine Kinase-dependent Suppression of a Voltage-gated Potassium Channel. Mol. Biol. Cell 2004, 15, 4073–4088. [Google Scholar] [CrossRef]
- Hattan, D.; Nesti, E.; Cachero, T.G.; Morielli, A.D. Tyrosine phosphorylation of Kv1.2 modulates its interaction with the actin-binding protein cortactin. J. Biol. Chem. 2002, 277, 38596–38606. [Google Scholar] [CrossRef] [Green Version]
- Bowlby, M.R.; Fadool, D.A.; Holmes, T.C.; Levitan, I.B. Modulation of the Kv1.3 Potassium Channel by Receptor Tyrosine Kinases. J. Gen. Physiol. 1997, 110, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Mármol, R.; Comes, N.; Styrczewska, K.; Pérez-Verdaguer, M.; Vicente, R.; Pujadas, L.; Soriano, E.; Sorkin, A.; Felipe, A. Unconventional EGF-induced ERK1/2-mediated Kv1.3 endocytosis. Cell. Mol. Life Sci. 2016, 73, 1515–1528. [Google Scholar] [CrossRef] [Green Version]
- Missan, S.; Qi, J.; Crack, J.; McDonald, T.F.; Linsdell, P. Regulation of wild-type and mutant KCNQ1/KCNE1 channels by tyrosine kinase. Pflug. Arch. 2009, 458, 471–480. [Google Scholar] [CrossRef]
- Kannankeril, P.; Roden, D.M.; Darbar, D. Drug-induced long QT syndrome. Pharm. Rev. 2010, 62, 760–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, A.T.; Nassal, D.; Deschenes, I.; Thomas, D.; Ficker, E. Antidepressant-induced ubiquitination and degradation of the cardiac potassium channel hERG. J. Biol. Chem. 2011, 286, 34413–34425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, S.M.; McEwen, D.P.; Zhang, L.; Arendt, K.L.; Van Genderen, K.M.; Martens, J.R. Antiarrhythmic drug-induced internalization of the atrial-specific k+ channel kv1.5. Circ. Res. 2009, 104, 1390–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Guo, J.; Shallow, H.; Yang, T.; Xu, J.; Li, W.; Hanson, C.; Wu, J.G.; Li, X.; Massaeli, H.; et al. The role of monoubiquitination in endocytic degradation of human ether-a-go-go-related gene (hERG) channels under low K+ conditions. J. Biol. Chem. 2011, 286, 6751–6759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaeli, H.; Sun, T.; Li, X.; Shallow, H.; Wu, J.; Xu, J.; Li, W.; Hanson, C.; Guo, J.; Zhang, S. Involvement of caveolin in low K+-induced endocytic degradation of cell-surface human ether-a-go-go-related gene (hERG) channels. J. Biol. Chem. 2010, 285, 27259–27264. [Google Scholar] [CrossRef] [Green Version]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Staub, O.; Abriel, H.; Plant, P.; Ishikawa, T.; Kanelis, V.; Saleki, R.; Horisberger, J.D.; Schild, L.; Rotin, D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000, 57, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Palmada, M.; Dieter, M.; Boehmer, C.; Waldegger, S.; Lang, F. Serum and glucocorticoid inducible kinases functionally regulate ClC-2 channels. Biochem. Biophys. Res. Commun. 2004, 321, 1001–1006. [Google Scholar] [CrossRef]
- Fotia, A.B.; Ekberg, J.; Adams, D.J.; Cook, D.I.; Poronnik, P.; Kumar, S. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J. Biol. Chem. 2004, 279, 28930–28935. [Google Scholar] [CrossRef] [Green Version]
- Henke, G.; Maier, G.; Wallisch, S.; Boehmer, C.; Lang, F. Regulation of the voltage gated K+ channel Kv1.3 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid inducible kinase SGK1. J. Cell. Physiol. 2004, 199, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, T.; Membrez, M.; Nicolas, C.S.; Pitard, B.; Staub, O.; Olesen, S.P.; Baro, I.; Abriel, H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res. 2007, 74, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Schuetz, F.; Boase, N.A.; Conroy, S.J.; Manning, J.; Kumar, S.; Poronnik, P.; Adams, D.J. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J. Biol. Chem. 2007, 282, 12135–12142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albesa, M.; Grilo, L.S.; Gavillet, B.; Abriel, H. Nedd4-2-dependent ubiquitylation and regulation of the cardiac potassium channel hERG1. J. Mol. Cell. Cardiol. 2011, 51, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ingham, R.J.; Gish, G.; Pawson, T. The Nedd4 family of E3 ubiquitin ligases: Functional diversity within a common modular architecture. Oncogene 2004, 23, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Abriel, H.; Loffing, J.; Rebhun, J.F.; Pratt, J.H.; Schild, L.; Horisberger, J.D.; Rotin, D.; Staub, O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J. Clin. Investig. 1999, 103, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Dibbens, L.M.; Ekberg, J.; Taylor, I.; Hodgson, B.L.; Conroy, S.J.; Lensink, I.L.; Kumar, S.; Zielinski, M.A.; Harkin, L.A.; Sutherland, G.R.; et al. NEDD4-2 as a potential candidate susceptibility gene for epileptic photosensitivity. Genesbrainand Behav. 2007, 6, 750–755. [Google Scholar] [CrossRef]
- Casini, S.; Albesa, M.; Wang, Z.; Portero, V.; Ross-Kaschitza, D.; Rougier, J.S.; Marchal, G.A.; Chung, W.K.; Bezzina, C.R.; Abriel, H.; et al. Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice. Int. J. Mol. Sci. 2019, 20, 33. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Krist, D.T.; Statsyuk, A.V. Crosstalk between kinases and Nedd4 family ubiquitin ligases. Mol. Biosyst. 2014, 10, 1643–1657. [Google Scholar] [CrossRef]
- Flores, S.Y.; Debonneville, C.; Staub, O. The role of Nedd4/Nedd4-like dependant ubiquitylation in epithelial transport processes. Pflug. Arch. 2003, 446, 334–338. [Google Scholar] [CrossRef]
- Wiemuth, D.; Ke, Y.; Rohlfs, M.; McDonald, F.J. Epithelial sodium channel (ENaC) is multi-ubiquitinated at the cell surface. Biochem. J. 2007, 405, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, P.M.; Olson, D.R.; Thomas, B.C. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J. Biol. Chem. 2002, 277, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laedermann, C.J.; Cachemaille, M.; Kirschmann, G.; Pertin, M.; Gosselin, R.D.; Chang, I.; Albesa, M.; Towne, C.; Schneider, B.L.; Kellenberger, S.; et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Investig. 2013, 123, 3002–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez, P.; Schwartz, A.B.; Iyer, S.R.; Warrington, A.; Fadool, D.A. Ubiquitin ligase Nedd4-2 modulates Kv1.3 current amplitude and ion channel protein targeting. J. Neurophysiol. 2016, 116, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Boehmer, C.; Laufer, J.; Jeyaraj, S.; Klaus, F.; Lindner, R.; Lang, F.; Palmada, M. Modulation of the voltage-gated potassium channel Kv1.5 by the SGK1 protein kinase involves inhibition of channel ubiquitination. Cell Physiol. Biochem. 2008, 22, 591–600. [Google Scholar] [CrossRef]
- MacGurn, J.A.; Hsu, P.C.; Emr, S.D. Ubiquitin and membrane protein turnover: From cradle to grave. Annu. Rev. Biochem. 2012, 81, 231–259. [Google Scholar] [CrossRef]
- Ye, S.; Cihil, K.; Stolz, D.B.; Pilewski, J.M.; Stanton, B.A.; Swiatecka-Urban, A. c-Cbl facilitates endocytosis and lysosomal degradation of cystic fibrosis transmembrane conductance regulator in human airway epithelial cells. J. Biol. Chem. 2010, 285, 27008–27018. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Fakitsas, P.; Adam, G.; Daidie, D.; van Bemmelen, M.X.; Fouladkou, F.; Patrignani, A.; Wagner, U.; Warth, R.; Camargo, S.M.; Staub, O.; et al. Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J. Am. Soc. Nephrol. 2007, 18, 1084–1092. [Google Scholar] [CrossRef] [Green Version]
- Ruffieux-Daidie, D.; Poirot, O.; Boulkroun, S.; Verrey, F.; Kellenberger, S.; Staub, O. Deubiquitylation regulates activation and proteolytic cleavage of ENaC. J. Am. Soc. Nephrol. 2008, 19, 2170–2180. [Google Scholar] [CrossRef]
- Krzystanek, K.; Rasmussen, H.B.; Grunnet, M.; Staub, O.; Olesen, S.P.; Abriel, H.; Jespersen, T. Deubiquitylating enzyme USP2 counteracts Nedd4-2-mediated downregulation of KCNQ1 potassium channels. Heart Rhythm 2012, 9, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Bomberger, J.M.; Barnaby, R.L.; Stanton, B.A. The deubiquitinating enzyme USP10 regulates the endocytic recycling of CFTR in airway epithelial cells. Channels (Austin) 2010, 4, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Attramadal, H.; Arriza, J.L.; Aoki, C.; Dawson, T.M.; Codina, J.; Kwatra, M.M.; Snyder, S.H.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [PubMed]
- Jean-Charles, P.Y.; Freedman, N.J.; Shenoy, S.K. Chapter Nine—Cellular Roles of Beta-Arrestins as Substrates and Adaptors of Ubiquitination and Deubiquitination. Prog. Mol. Biol. Transl. Sci. 2016, 141, 339–369. [Google Scholar] [CrossRef] [PubMed]
- Claing, A.; Laporte, S.A.; Caron, M.G.; Lefkowitz, R.J. Endocytosis of G protein-coupled receptors: Roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog. Neurobiol. 2002, 66, 61–79. [Google Scholar] [CrossRef]
- Nagi, K.; Charfi, I.; Pineyro, G. Kir3 channels undergo arrestin-dependant internalization following delta opioid receptor activation. Cell Mol. Life Sci. 2015, 72, 3543–3557. [Google Scholar] [CrossRef]
- Hermosilla, T.; Encina, M.; Morales, D.; Moreno, C.; Conejeros, C.; Alfaro-Valdes, H.M.; Lagos-Meza, F.; Simon, F.; Altier, C.; Varela, D. Prolonged AT1R activation induces CaV1.2 channel internalization in rat cardiomyocytes. Sci. Rep. 2017, 7, 10131. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Kim, J.; Ahn, S.; Xiao, K.; Shenoy, S.K.; Liedtke, W.; Lefkowitz, R.J. Arresting a transient receptor potential (TRP) channel: Beta-arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4. J. Biol. Chem. 2010, 285, 30115–30125. [Google Scholar] [CrossRef] [Green Version]
- Simonin, A.; Fuster, D. Nedd4-1 and beta-arrestin-1 are key regulators of Na+/H+ exchanger 1 ubiquitylation, endocytosis, and function. J. Biol. Chem. 2010, 285, 38293–38303. [Google Scholar] [CrossRef] [Green Version]
- Somsel Rodman, J.; Wandinger-Ness, A. Rab GTPases coordinate endocytosis. J. Cell Sci. 2000, 113 Pt 2, 183–192. [Google Scholar]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluijs, P.; Hull, M.; Webster, P.; Male, P.; Goud, B.; Mellman, I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 1992, 70, 729–740. [Google Scholar] [CrossRef]
- Zadeh, A.D.; Xu, H.; Loewen, M.E.; Noble, G.P.; Steele, D.F.; Fedida, D. Internalized Kv1.5 traffics via Rab-dependent pathways. J. Physiol. 2008, 586, 4793–4813. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Fast endocytic recycling determines TRPC1–STIM1 clustering in ER–PM junctions and plasma membrane function of the channel. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2709–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Zhang, S. Regulation of the human ether-a-go-go-related gene (hERG) channel by Rab4 protein through neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2). J. Biol. Chem. 2013, 288, 21876–21886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; Matos, P. Rab GTPases regulate the trafficking of channels and transporters—A focus on cystic fibrosis. Small Gtpases 2018, 9, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Gorvel, J.P.; Chavrier, P.; Zerial, M.; Gruenberg, J. rab5 controls early endosome fusion in vitro. Cell 1991, 64, 915–925. [Google Scholar] [CrossRef]
- Seebohm, G.; Strutz-Seebohm, N.; Birkin, R.; Dell, G.; Bucci, C.; Spinosa, M.R.; Baltaev, R.; Mack, A.F.; Korniychuk, G.; Choudhury, A.; et al. Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ. Res. 2007, 100, 686–692. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Press, B.; Wandinger-Ness, A. Rab 7: An important regulator of late endocytic membrane traffic. J. Cell Biol. 1995, 131 Pt 1, 1435–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, O.; Reinsch, S.; Urbe, S.; Zerial, M.; Parton, R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996, 135, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, Y.; Chen, D.; Wandinger-Ness, A. Rab11 is required for trans-golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol. Biol. Cell 1998, 9, 3241–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guo, J.; Yang, T.; Li, W.; Lamothe, S.M.; Kang, Y.; Szendrey, J.A.; Zhang, S. Rab11-dependent Recycling of the Human Ether-a-go-go-related Gene (hERG) Channel. J. Biol. Chem. 2015, 290, 21101–21113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpushev, A.V.; Levchenko, V.; Pavlov, T.S.; Lam, V.; Vinnakota, K.C.; Vandewalle, A.; Wakatsuki, T.; Staruschenko, A. Regulation of ENaC expression at the cell surface by Rab11. Biochem. Biophys. Res. Commun. 2008, 377, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Van de Graaf, S.F.; Chang, Q.; Mensenkamp, A.R.; Hoenderop, J.G.; Bindels, R.J. Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane. Mol. Cell. Biol. 2006, 26, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Gwozdzinska, P.; Buchbinder, B.A.; Mayer, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadasz, I. Hypercapnia Impairs ENaC Cell Surface Stability by Promoting Phosphorylation, Polyubiquitination and Endocytosis of beta-ENaC in a Human Alveolar Epithelial Cell Line. Front. Immunol. 2017, 8, 591. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.K.; Singh, M.; Shibata, H.; Kaur, S.; George, C. Rab4 GTP/GDP modulates amiloride-sensitive sodium channel (ENaC) function in colonic epithelia. Biochem. Biophys. Res. Commun. 2006, 340, 726–733. [Google Scholar] [CrossRef]
- Huang, X.Y.; Morielli, A.D.; Peralta, E.G. Tyrosine kinase-dependent suppression of a potassium channel by the G protein-coupled m1 muscarinic acetylcholine receptor. Cell 1993, 75, 1145–1156. [Google Scholar] [CrossRef]
- Tsai, W.; Morielli, A.D.; Peralta, E.G. The m1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. Embo J. 1997, 16, 4597–4605. [Google Scholar] [CrossRef] [Green Version]
- Cogolludo, A.; Moreno, L.; Lodi, F.; Frazziano, G.; Cobeno, L.; Tamargo, J.; Perez-Vizcaino, F. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: Role of 5-HT2A receptors, caveolin-1, and KV1.5 channel internalization. Circ. Res. 2006, 98, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.N.; Skibsbye, L.; Tang, C.; Petersen, F.; MacAulay, N.; Rasmussen, H.B.; Jespersen, T. PKC and AMPK regulation of Kv1.5 potassium channels. Channels (Austin) 2015, 9, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, V.A.; Purtell, K.; Abbott, G.W. Protein Kinase C downregulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm Off. J. Heart Rhythm Soc. 2011, 8, 1641–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebohm, G.; Strutz-Seebohm, N.; Ureche, O.N.; Henrion, U.; Baltaev, R.; Mack, A.F.; Korniychuk, G.; Steinke, K.; Tapken, D.; Pfeufer, A.; et al. Long QT syndrome-associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ. Res. 2008, 103, 1451–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benned-Jensen, T.; Christensen, R.K.; Denti, F.; Perrier, J.F.; Rasmussen, H.B.; Olesen, S.P. Live Imaging of Kv7.2/7.3 Cell Surface Dynamics at the Axon Initial Segment: High Steady-State Stability and Calpain-Dependent Excitotoxic Downregulation Revealed. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 2261–2266. [Google Scholar]
- Anta, B.; Martin-Rodriguez, C.; Gomis-Perez, C.; Calvo, L.; Lopez-Benito, S.; Calderon-Garcia, A.A.; Vicente-Garcia, C.; Villarroel, A.; Arevalo, J.C. Ubiquitin-specific Protease 36 (USP36) Controls Neuronal Precursor Cell-expressed Developmentally Down-regulated 4-2 (Nedd4-2) Actions over the Neurotrophin Receptor TrkA and Potassium Voltage-gated Channels 7.2/3 (Kv7.2/3). J. Biol. Chem. 2016, 291, 19132–19145. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.H.; Sterling, H.; Wang, Z.; Babilonia, E.; Yang, B.; Dong, K.; Hebert, S.C.; Giebisch, G.; Wang, W.H. ROMK1 channel activity is regulated by monoubiquitination. Proc. Natl. Acad. Sci. USA 2005, 102, 4306–4311. [Google Scholar] [CrossRef] [Green Version]
- Ronzaud, C.; Loffing-Cueni, D.; Hausel, P.; Debonneville, A.; Malsure, S.R.; Fowler-Jaeger, N.; Boase, N.A.; Perrier, R.; Maillard, M.; Yang, B.; et al. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J. Clin. Investig. 2013, 123, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.H.; Yue, P.; Pan, C.Y.; Sun, P.; Zhang, X.; Han, Z.; Roos, M.; Caplan, M.; Giebisch, G.; Wang, W.H. POSH stimulates the ubiquitination and the clathrin-independent endocytosis of ROMK1 channels. J. Biol. Chem. 2009, 284, 29614–29624. [Google Scholar] [CrossRef] [Green Version]
- Alewine, C.; Kim, B.Y.; Hegde, V.; Welling, P.A. Lin-7 targets the Kir 2.3 channel on the basolateral membrane via a L27 domain interaction with CASK. Am. J. Physiol. Cell Physiol. 2007, 293, C1733–C1741. [Google Scholar] [CrossRef]
- Ma, D.; Zerangue, N.; Raab-Graham, K.; Fried, S.R.; Jan, Y.N.; Jan, L.Y. Diverse Trafficking Patterns Due to Multiple Traffic Motifs in G Protein-Activated Inwardly Rectifying Potassium Channels from Brain and Heart. Neuron 2002, 33, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Huang, C.S.; Jan, Y.N.; Jan, L.Y. ATP-Sensitive Potassium Channel Traffic Regulation by Adenosine and Protein Kinase, C. Neuron 2003, 38, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Miake, J.; Notsu, T.; Sonyama, K.; Sasaki, N.; Iitsuka, K.; Kato, M.; Taniguchi, S.; Igawa, O.; Yoshida, A.; et al. Proteasomal degradation of Kir6.2 channel protein and its inhibition by a Na+ channel blocker aprindine. Biochem. Biophys. Res. Commun. 2005, 331, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, K.; Chapline, C.; Sato, J.D.; Stanton, B.A. Nedd4-2 does not regulate wt-CFTR in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L720–L727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhani, S.U.; Kim Chiaw, P.; Huan, L.-J.; Bear, C.E. ATP depletion inhibits the endocytosis of ClC-2. J. Cell. Physiol. 2008, 214, 273–280. [Google Scholar] [CrossRef]
- Zhang, W.; Na, T.; Wu, G.; Jing, H.; Peng, J.-B. Down-regulation of intestinal apical calcium entry channel TRPV6 by ubiquitin E3 ligase Nedd4-2. J. Biol. Chem. 2010, 285, 36586–36596. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-J.; Zhu, B.-L.; Sun, F.; Luo, D.; Ma, Y.-L.; Luo, B.; Tang, J.; Xiong, M.-J.; Liu, L.; Long, Y.; et al. Estrogen receptor α promotes Cav1.2 ubiquitination and degradation in neuronal cells and in APP/PS1 mice. Aging Cell 2019, 18, e12961. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ion Channel | Type of Endocytosis | Stimulus of Internalization | Ubiquitination | Rabs | Associated Pathologies |
|---|---|---|---|---|---|
| ENaC | CME [12] | Kidney: Basal conditions (low aldosterone) [75] Alveoli: hypercapnia [110] | Polyubiquitination [74] E3 Nedd4-2 [75] DUB USP2-45 [82,83] | Rab4 [111] Rab11 [108] | Liddle’s syndrome [69] |
| Nav family (Nav1.1-1.3 and 1.5-1.8) | ND | ND | Polyubiquitination E3 Nedd4-2 [63] | ND | Congenital type 3 long QT syndrome # (Nav1.5) [71] Neuropathic pain (Nav1.7-1.8) [76] |
| Kv1.2 | CME, RhoA and cholesterol-dependent [45] | Constitutive [45] and Receptor-mediated: mAChR [112] and EGFR [113] | ND | ND | ND |
| Kv1.3 | CME [13,52] | Receptor-mediated (EGFR) [52] and PKC [13] | Polyubiquitination E3 Nedd4-2 * [13,64,77] | ND | ND |
| Kv1.5 | CME ‡ [97] Caveolin-mediated [114] | Receptor-mediated (5-HT) [114], Drug-induced (quinidine) [56], PKC and AMPK [115] | Ubiquitination E3 Nedd4-2 * [78] | Rab4, Rab5, Rab7, Rab11 [97] | ND |
| Kv7.1 | CME [14] | Receptor-mediated (α1AR) [14] and PKC [116] | Polyubiquitination E3 Nedd4-2 [14,65] DUB USP2-45 and USP2-63 [84] | Rab5, Rab11 [103] | Type 1 long QT syndrome [117] |
| Kv7.2/7.3 | CME [118] | Receptor and calpain-mediated (NMDA—high glutamate) [118] | Polyubiquitination E3 Nedd4-2 [66] DUB USP36 [119] | ND | ND |
| Kv11.1 | Caveolin [57,58] ARF6-mediated [48] | Drug (Desipramine) [55] and Low K+-induced [57] | Monoubiquitination E3 Nedd4-2 [67] | Rab4 [99] Rab11 [107] | Acquired long QT syndrome [55] |
| Kir1.1 (ROMK) | CME [24] Caveolin-mediated [44] | Constitutive [24], WNK [24] and PTK kinases [120] | Monoubiquitination [120] E3 Nedd4-2 * [121] CIE: E3 POSH [122] | ND | ND |
| Kir2.3 | CME [17] | Constitutive [17] | ND | Rab11 [123] | ND |
| Kir3.1/3.2 | CME [90] | Receptor-mediated (DOR) [90] | ND | Rab7 [124] | ND |
| Kir3.4 | ARF6-mediated [46] | ND | ND | Rab7 [124] | ND |
| Kir6.1 | Caveolin-mediated [40] | PKC [40] | ND | ND | ND |
| Kir6.2 | CME [7] | PKC [125] | Ubiquitination [126] | Rab7 [125] | Permanent neonatal diabetes mellitus [7] |
| CFTR | CME [15] | ND | Polyubiqutination E3 CHIP [127], c-Cbl [80] and Nedd4-2 * (ΔF508) [128] DUB USP10 [85] | Rab4, Rab5, Rab7, Rab11 [100] | ND |
| CLC-2 | Dynamin-dependent [129] | ND | Polyubiquitination E3 Nedd4-2 * [62] | Rab5, Rab11 [129] | ND |
| TRPC1 | ARF6-mediated [98] | ND | ND | Rab4, Rab5 [98] | ND |
| TRPV4 | CME ‡ [92] | Receptor-mediated (AT1R) [92] | Ubiquitination E3 AIP4 [92] | ND | ND |
| TRPV5 | CME [42] Caveolin-mediated [41] | Constitutive [41] | E3 Nedd4-2 * [130] | Rab11 [42] | ND |
| Cav1.2 | CME [16] | Receptor-mediated: AT1R [91] and ERα [131] | Polyubiquitination E3 Mdm2 [131] | Rab11 [16] | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estadella, I.; Pedrós-Gámez, O.; Colomer-Molera, M.; Bosch, M.; Sorkin, A.; Felipe, A. Endocytosis: A Turnover Mechanism Controlling Ion Channel Function. Cells 2020, 9, 1833. https://doi.org/10.3390/cells9081833
Estadella I, Pedrós-Gámez O, Colomer-Molera M, Bosch M, Sorkin A, Felipe A. Endocytosis: A Turnover Mechanism Controlling Ion Channel Function. Cells. 2020; 9(8):1833. https://doi.org/10.3390/cells9081833
Chicago/Turabian StyleEstadella, Irene, Oriol Pedrós-Gámez, Magalí Colomer-Molera, Manel Bosch, Alexander Sorkin, and Antonio Felipe. 2020. "Endocytosis: A Turnover Mechanism Controlling Ion Channel Function" Cells 9, no. 8: 1833. https://doi.org/10.3390/cells9081833
APA StyleEstadella, I., Pedrós-Gámez, O., Colomer-Molera, M., Bosch, M., Sorkin, A., & Felipe, A. (2020). Endocytosis: A Turnover Mechanism Controlling Ion Channel Function. Cells, 9(8), 1833. https://doi.org/10.3390/cells9081833