Discovery of Molecular DNA Methylation-Based Biomarkers through Genome-Wide Analysis of Response Patterns to BCG for Bladder Cancer

 , , ,
, , ,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Tissue Samples
2.2. DNA Extraction and Bisulfite Conversion
2.3. Epigenome-Wide DNA Methylation Analysis with ILLUMINA’S Infinium MethylationEPIC BeadChips
2.4. Data and Pathway Analysis
2.5. Targeted Bisulfite Sequencing
2.6. Statistical Evaluation of Targeted Bisulfite Sequencing
2.7. Immunohistochemistry
3. Results
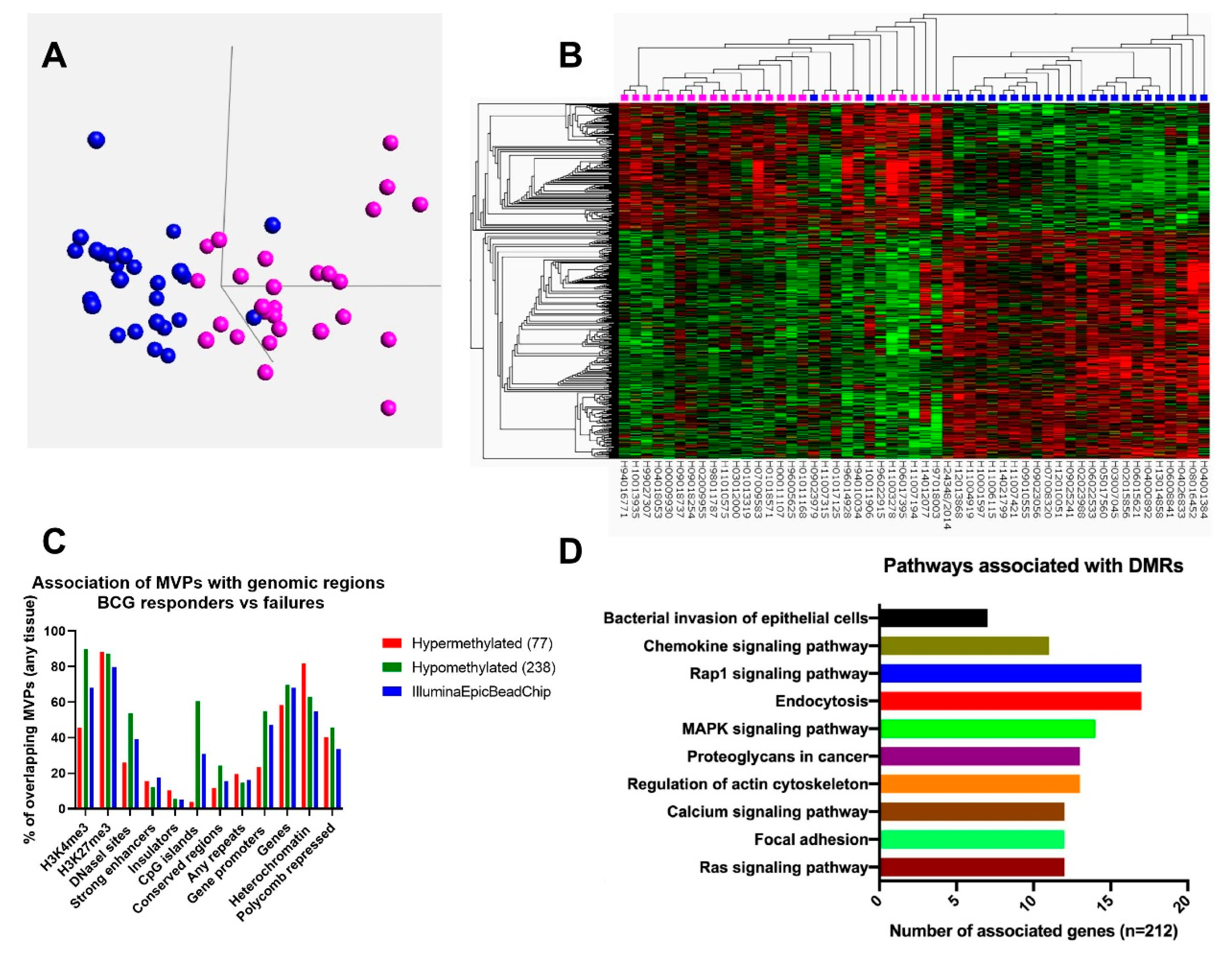
3.1. Genome Wide DNA Methylation Analysis Reveals Distinct CpG Methylation Patterns for BCG Responders and Failures
3.2. BCG Failures Show More pronounced Gain and Loss of DNA Methylation at Sites Known for Cancer-Specific Alterations
3.3. Differentially Methylated Regions between BCG Responders and Failures are Associated with Genes Involved in Bacterial Uptake and Cell-Adhesion Pathways
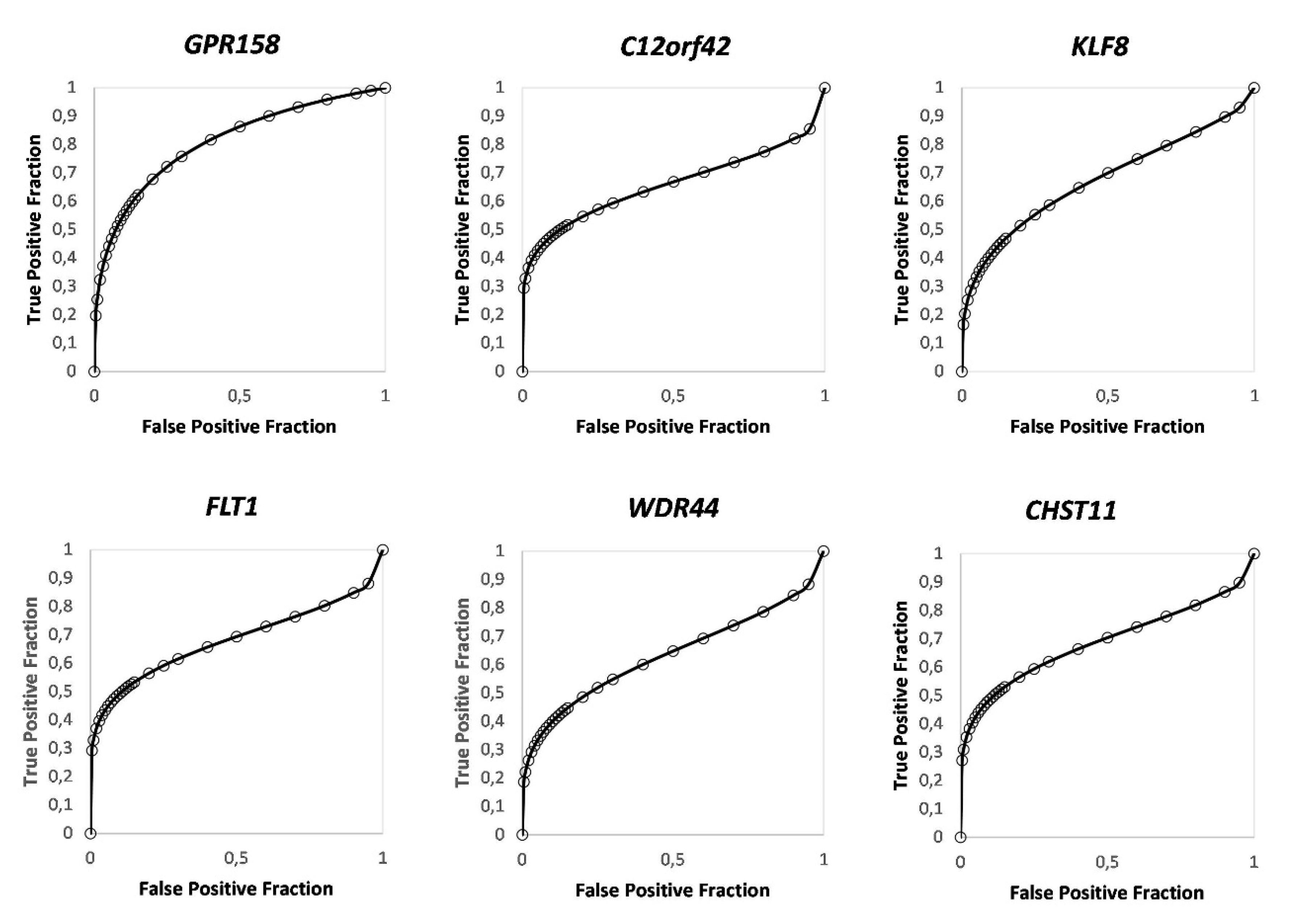
3.4. Identification of Candidate DNA Methylation Biomarkers for BCG Response
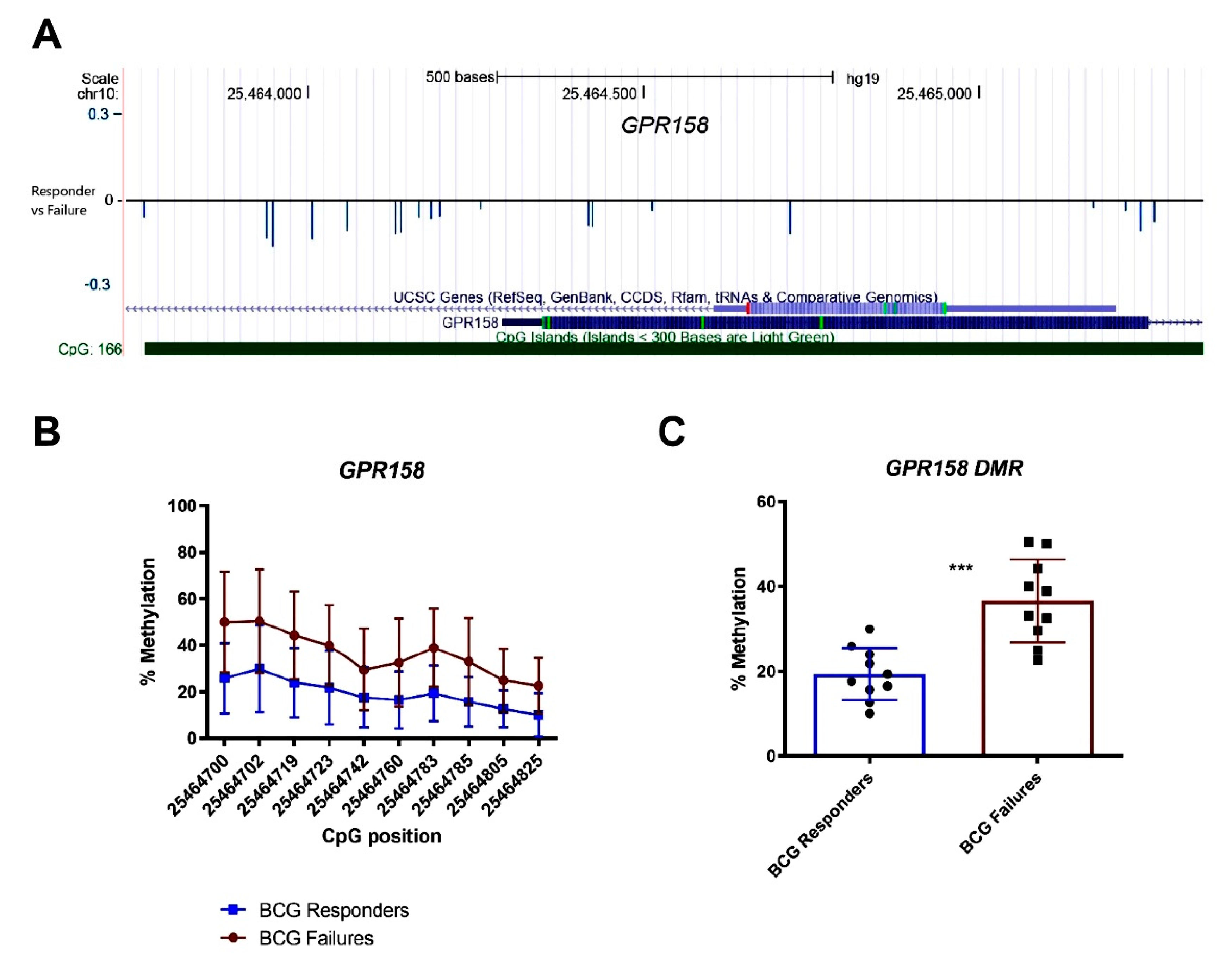
3.5. GRP158 Shows a Higher Degree of Methylation in Patients with BCG Failure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; Van Rhijn, B.W.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma In Situ)—2019 Update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef] [PubMed]
- Böhle, A.; Bock, P. Intravesical bacille calmette-guérin versus mitomycin c in superficial bladder cancer: Formal meta-analysis of comparative studies on tumor progression. Urology 2004, 63, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, R.J.; Van Der Meijden, A.P.M.; Lamm, D.L. Intravesical bacillus Calmette-Guerin reduces the risk of progression in patients with superficial bladder cancer: A meta-analysis of the published results of randomized clinical trials. J. Urol. 2002, 168, 1964–1970. [Google Scholar] [CrossRef]
- Xylinas, E.; Kent, M.; Sjoberg, D.; Kluth, L.; Pycha, A.; Comploj, E.; Lotan, Y.; Svatek, R.; Trinh, Q.-D.; Karakiewicz, P.; et al. 1701 Accuracy of the Eortc Risk Tables and of the Cueto Scoring Model to Predict Outcomes in Non Muscle-Invasive Urothelial Carcinoma of the Bladder. J. Urol. 2013, 189, 1460–1466. [Google Scholar] [CrossRef] [Green Version]
- Gontero, P.; Sylvester, R.; Pisano, F.; Joniau, S.; Eeckt, K.V.; Serretta, V.; Larré, S.; Di Stasi, S.; Van Rhijn, B.; Witjes, A.J.; et al. Prognostic Factors and Risk Groups in T1G3 Non–Muscle-invasive Bladder Cancer Patients Initially Treated with Bacillus Calmette-Guérin: Results of a Retrospective Multicenter Study of 2451 Patients. Eur. Urol. 2015, 67, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Pettenati, C.; Ingersoll, M.A. Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat. Rev. Urol. 2018, 15, 615–625. [Google Scholar] [CrossRef]
- Bisiaux, A.; Thiounn, N.; Timsit, M.-O.; Eladaoui, A.; Chang, H.-H.; Mapes, J.; Mogenet, A.; Bresson, J.-L.; Prié, D.; Béchet, S.; et al. Molecular Analyte Profiling of the Early Events and Tissue Conditioning Following Intravesical Bacillus Calmette-Guerin Therapy in Patients With Superficial Bladder Cancer. J. Urol. 2009, 181, 1571–1580. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, X.; O’Donnell, M. Role of Th1 and Th2 cytokines in BCG-induced IFN-gamma production: Cytokine promotion and simulation of BCG effect. Cytokine 2003, 21, 17–26. [Google Scholar] [CrossRef]
- De Boer, E.C.; De Jong, W.H.; Steerenberg, P.A.; Aarden, L.A.; Tetteroo, E.; De Groot, E.R.; Van Der Meijden, A.P.M.; Vegt, P.D.J.; Debruyne, F.M.J.; Ruitenberg, E.J. Induction of urinary interleukin-1 (IL-1), IL-2, IL-6, and tumour necrosis factor during intravesical immunotherapy with bacillus Calmette-Guérin in superficial bladder cancer. Cancer Immunol. Immunother. 1992, 34, 306–312. [Google Scholar] [CrossRef]
- Böhle, A.; Brandau, S. Immune Mechanisms in Bacillus Calmette-Guerin Immunotherapy for Superficial Bladder Cancer. J. Urol. 2003, 170, 964–969. [Google Scholar] [CrossRef]
- Teppema, J.S.; De Boer, E.C.; Steerenberg, P.A.; Van Der Meijden, A.P.M. Morphological aspects of the interaction of Bacillus Calmette-Guérin with urothelial bladder cells in vivo and in vitro: Relevance for antitumor activity? Urol. Res. 1992, 20, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Brown, E.J.; Telle, W.B.; Russell, D.G.; Ratliff, T.L. Characterization of the internalization of bacillus Calmette-Guerin by human bladder tumor cells. J. Clin. Investig. 1993, 91, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Kavoussi, L.R.; Brown, E.J.; Ritchey, J.K.; Ratliff, T.L. Fibronectin-mediated Calmette-Guerin bacillus attachment to murine bladder mucosa. Requirement for the expression of an antitumor response. J. Clin. Investig. 1990, 85, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redelman-Sidi, G.; Iyer, G.; Solit, D.B.; Glickman, M.S. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res. 2013, 73, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Durek, C.; Brandau, S.; Ulmer, A.J.; Flad, H.-D.; Jocham, D.; Bohle, A. Bacillus-Calmette-Guerin (BCG) and 3D Tumors: An in vitro Model for the Study of Adhesion and Invasion. J. Urol. 1999, 162, 600–605. [Google Scholar] [CrossRef]
- Becich, M.J.; Carroll, S.; Ratliff, T.L. Internalization of Bacille Calmette-Guerin by Bladder Tumor Cells. J. Urol. 1991, 145, 1316–1324. [Google Scholar] [CrossRef]
- Pook, S.-H.; Rahmat, J.; Esuvaranathan, K.; Mahendran, R. Internalization of Mycobacterium bovis, Bacillus Calmette Guerin, by bladder cancer cells is cytotoxic. Oncol. Rep. 2007, 18, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, N.; Toida, I.; Iwasaki, A.; Kawai, K.; Akaza, H. Surface antigen expression on bladder tumor cells induced by bacillus Calmette-Guérin (BCG): A role of BCG internalization into tumor cells. Int. J. Urol. 2002, 9, 29–35. [Google Scholar] [CrossRef]
- Stefanini, G.; Bercovich, E.; Mazzeo, V.; Grigioni, W.; Emili, E.; D’Errico, A.; Cigno, M.L.; Tamagnini, N.; Mazzetti, M. Class I and Class II HLA Antigen Expression by Transitional Cell Carcinoma of the Bladder: Correlation with T-Cell Infiltration and BCG Treatment. J. Urol. 1989, 141, 1449–1453. [Google Scholar] [CrossRef]
- Suttmann, H.; Riemensberger, J.; Bentien, G.; Schmaltz, D.; Stöckle, M.; Jocham, D.; Böhle, A.; Brandau, S. Neutrophil Granulocytes Are Required for EffectiveBacillus Calmette-GuérinImmunotherapy of Bladder Cancer and Orchestrate Local Immune Responses. Cancer Res. 2006, 66, 8250–8257. [Google Scholar] [CrossRef] [Green Version]
- Ilijazi, D.; Abufaraj, M.; Hassler, M.R.; Ertl, I.E.; D’Andrea, D.; Shariat, S.F. Waiting in the wings: The emerging role of molecular biomarkers in bladder cancer. Expert Rev. Mol. Diagn. 2018, 18, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Múgica, M.; Fernandez-Gomez, J.M.; Cebrian, V.; Fresno, F.; Escaf, S.; Sanchez-Carbayo, M. Polyamine-modulated Factor-1 Methylation Predicts Bacillus Calmette-Guérin Response in Patients with High-grade Non–muscle-invasive Bladder Carcinoma. Eur. Urol. 2013, 63, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, M.; Bryan, R.T.; Emes, R.D.; Luscombe, C.J.; Cheng, K.; Zeegers, M.P.; James, N.D.; Gommersall, L.M.; Fryer, A. HumanMethylation450K Array–Identified Biomarkers Predict Tumour Recurrence/Progression at Initial Diagnosis of High-risk Non-muscle Invasive Bladder Cancer. Biomarkers Cancer 2018, 10, 1179299–17751920. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.; Molloy, P.L.; Van Dijk, S.J.; Muhlhausler, B.S.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Boil. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Metcalfe, M.; Tabayoyong, W.B.; Guo, C.; González, G.M.N.; Navai, N.; Grossman, H.B.; Dinney, C.P.N.; Kamat, A.M. Using Grade of Recurrent Tumor to Guide Further Therapy While on Bacillus Calmette-Guerin: Low-grade Recurrences Are not Benign. Eur. Urol. Oncol. 2018, 2, 286–293. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Zhang, Y.; Storey, J.D.; Torres, L.C. Bioconductor – sva: Surrogate Variable Analysis. Available online: https://bioconductor.org/packages/release/bioc/html/sva.html (accessed on 31 July 2020).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Murakami, P.; Lee, H.; Leek, J.T.; Fallin, M.D.; Feinberg, A.P.; Irizarry, R.A. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 2012, 41, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Halachev, K.; Bast, H.; Albrecht, F.; Lengauer, T.; Bock, C. EpiExplorer: Live exploration and global analysis of large epigenomic datasets. Genome Boil. 2012, 13, R96. [Google Scholar] [CrossRef] [Green Version]
- Pabinger, S.; Ernst, K.; Pulverer, W.; Kallmeyer, R.; Valdes, A.M.; Metrustry, S.; Katic, D.; Nuzzo, A.; Kriegner, A.; Vierlinger, K.; et al. Analysis and Visualization Tool for Targeted Amplicon Bisulfite Sequencing on Ion Torrent Sequencers. PLoS ONE 2016, 11, e0160227. [Google Scholar] [CrossRef]
- Simon, R.; Lam, A.; Li, M.-C.; Ngan, M.; Menenzes, S.; Zhao, Y. Analysis of Gene Expression Data Using BRB-Array Tools. Cancer Inform. 2007, 3, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, d.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-Y.; Sun, K.-H.; Chen, S.-Y.; Wang, H.-H.; Lee, M.-Y.; Tsou, Y.-C.; Jwo, S.-C.; Sun, G.-H.; Tang, S.-J. Autocrine CCL2 promotes cell migration and invasion via PKC activation and tyrosine phosphorylation of paxillin in bladder cancer cells. Cytokine 2012, 59, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Petit, V.; Boyer, B.; Lentz, D.; Turner, C.E.; Thiery, J.P.; Vallés, A.M. Phosphorylation of Tyrosine Residues 31 and 118 on Paxillin Regulates Cell Migration through an Association with Crk in Nbt-II Cells. J. Cell Boil. 2000, 148, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Metalli, D.; Lovat, F.; Tripodi, F.; Genua, M.; Xu, S.-Q.; Spinelli, M.; Alberghina, F.; Vanoni, M.; Baffa, R.; Gomella, L.G.; et al. The Insulin-Like Growth Factor Receptor I Promotes Motility and Invasion of Bladder Cancer Cells through Akt- and Mitogen-Activated Protein Kinase-Dependent Activation of Paxillin. Am. J. Pathol. 2010, 176, 2997–3006. [Google Scholar] [CrossRef]
- Babjuk, M.; Böhle, A.; Burger, M.; Čapoun, O.; Cohen, D.; Compérat, E.; Hernández, V.; Kaasinen, E.; Palou, J.; Rouprêt, M.; et al. EAU Guidelines on Non–Muscle-invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur. Urol. 2017, 71, 447–461. [Google Scholar] [CrossRef]
- Rechache, N.S.; Wang, Y.; Stevenson, H.S.; Killian, J.K.; Edelman, D.C.; Merino, M.; Zhang, L.; Nilubol, N.; Stratakis, C.A.; Meltzer, P.S.; et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J. Clin. Endocrinol. Metab. 2012, 97, E1004–E1013. [Google Scholar] [CrossRef] [Green Version]
- Geybels, M.S.; Zhao, S.; Wong, C.-J.; Bibikova, M.; Klotzle, B.; Wu, M.C.; Ostrander, E.A.; Fan, J.-B.; Feng, Z.; Stanford, J.L. Epigenomic profiling of DNA methylation in paired prostate cancer versus adjacent benign tissue. Prostate 2015, 75, 1941–1950. [Google Scholar] [CrossRef] [Green Version]
- Olkhov-Mitsel, E.; Savio, A.J.; Kron, K.J.; Pethe, V.V.; Hermanns, T.; Fleshner, N.E.; Van Rhijn, B.; Van Der Kwast, T.H.; Zlotta, A.R.; Bapat, B. Epigenome-Wide DNA Methylation Profiling Identifies Differential Methylation Biomarkers in High-Grade Bladder Cancer. Transl. Oncol. 2017, 10, 168–177. [Google Scholar] [CrossRef]
- Smith, B.A.; Balanis, N.G.; Nanjundiah, A.; Sheu, K.M.; Tsai, B.; Zhang, Q.; Park, J.W.; Thompson, M.; Huang, J.; Witte, O.N.; et al. A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep. 2018, 24, 3353–3366.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monami, G.; Gonzalez, E.M.; Hellman, M.; Gomella, L.G.; Baffa, R.; Iozzo, R.V.; Morrione, A. Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex. Cancer Res. 2006, 66, 7103–7110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasopoulou, A.; Aroukatos, P.; Nakas, D.; Repanti, M.; Papadaki, H.; Bravou, V. Decreased ezrin and paxillin expression in human urothelial bladder tumors correlate with tumor progression. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inform. 2019, 18, 1–11. [Google Scholar] [CrossRef]
- Orlandi, C.; Xie, K.; Masuho, I.; Fajardo-Serrano, A.; Lujan, R.; Martemyanov, K.A. Orphan Receptor GPR158 Is an Allosteric Modulator of RGS7 Catalytic Activity with an Essential Role in Dictating Its Expression and Localization in the Brain. J. Boil. Chem. 2015, 290, 13622–13639. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Itakura, T.; González, J.M.; Schwartz, S.G.; Fini, M.E. GPR158, an Orphan Member of G Protein-Coupled Receptor Family C: Glucocorticoid-Stimulated Expression and Novel Nuclear Role. PLoS ONE 2013, 8, e57843. [Google Scholar] [CrossRef]
- Patel, N.; Itakura, T.; Jeong, S.; Liao, C.-P.; Roy-Burman, P.; Zandi, E.; Groshen, S.; Pinski, J.; Coetzee, G.A.; Gross, M.E.; et al. Expression and Functional Role of Orphan Receptor GPR158 in Prostate Cancer Growth and Progression. PLoS ONE 2015, 10, e0117758. [Google Scholar] [CrossRef]
- Kitchen, M.; Bryan, R.T.; Haworth, K.E.; Emes, R.D.; Luscombe, C.; Gommersall, L.; Cheng, K.K.; Zeegers, M.P.; James, N.D.; Devall, A.J.; et al. Methylation of HOXA9 and ISL1 Predicts Patient Outcome in High-Grade Non-Invasive Bladder Cancer. PLoS ONE 2015, 10, e0137003. [Google Scholar] [CrossRef]
- Agundez, M.; Grau, L.; Palou, J.; Algaba, F.; Villavicencio, H.; Sanchez-Carbayo, M. Evaluation of the Methylation Status of Tumour Suppressor Genes for Predicting Bacillus Calmette-Guérin Response in Patients With T1G3 High-Risk Bladder Tumours. Eur. Urol. 2011, 60, 131–140. [Google Scholar] [CrossRef]
- Heiss, J.A.; Brennan, K.J.; Baccarelli, A.A.; Téllez-Rojo, M.M.; Estrada-Gutierrez, G.; Wright, R.O.; Just, A.C. Battle of epigenetic proportions: Comparing Illumina’s EPIC methylation microarrays and TruSeq targeted bisulfite sequencing. Epigenetics 2019, 15, 174–182. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic 1 | Responders (n = 26) | Failures (n = 27) |
|---|---|---|
| Age, y | ||
| Median | 69 | 70 |
| Range | 51–86 | 54–93 |
| Sex, no. (%) | ||
| Male | 23 (88) | 20 (74) |
| Female | 3 (12) | 7 (26) |
| T stage, no. (%) | ||
| Tis | 0 | 1 (4) |
| Ta | 7 (27) | 2 (8) |
| T1 | 19 (73) | 24 (88) |
| T1a | 10 (38) | 8 (30) |
| T1b | 6 (23) | 4 (14) |
| T1 ns | 3 (12) | 12 (44) |
| Grade, no. (%) | ||
| High | 24 (92) | 24 (89) |
| Not specified | 2 (8) | 3 (11) |
| Concomitant CIS, no. (%) | 16 (62) | 11 (41) |
| Type of failure, no. (%) | ||
| BCG refr. + early rec. | - | 18 (67) |
| BCG late rec. | - | 9 (33) |
| Follow-up since TURB, m | 26 (5–109) | 12 (2–38) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilijazi, D.; Pulverer, W.; Ertl, I.E.; Lemberger, U.; Kimura, S.; Abufaraj, M.; D’Andrea, D.; Pradere, B.; Bruchbacher, A.; Graf, A.; et al. Discovery of Molecular DNA Methylation-Based Biomarkers through Genome-Wide Analysis of Response Patterns to BCG for Bladder Cancer. Cells 2020, 9, 1839. https://doi.org/10.3390/cells9081839
Ilijazi D, Pulverer W, Ertl IE, Lemberger U, Kimura S, Abufaraj M, D’Andrea D, Pradere B, Bruchbacher A, Graf A, et al. Discovery of Molecular DNA Methylation-Based Biomarkers through Genome-Wide Analysis of Response Patterns to BCG for Bladder Cancer. Cells. 2020; 9(8):1839. https://doi.org/10.3390/cells9081839
Chicago/Turabian StyleIlijazi, Dafina, Walter Pulverer, Iris E. Ertl, Ursula Lemberger, Shoji Kimura, Mohammad Abufaraj, David D’Andrea, Benjamin Pradere, Andreas Bruchbacher, Anna Graf, and et al. 2020. "Discovery of Molecular DNA Methylation-Based Biomarkers through Genome-Wide Analysis of Response Patterns to BCG for Bladder Cancer" Cells 9, no. 8: 1839. https://doi.org/10.3390/cells9081839
APA StyleIlijazi, D., Pulverer, W., Ertl, I. E., Lemberger, U., Kimura, S., Abufaraj, M., D’Andrea, D., Pradere, B., Bruchbacher, A., Graf, A., Soria, F., Susani, M., Haitel, A., Molinaro, L., Pycha, A., Comploj, E., Pabinger, S., Weinhäusel, A., Egger, G., ... Hassler, M. R. (2020). Discovery of Molecular DNA Methylation-Based Biomarkers through Genome-Wide Analysis of Response Patterns to BCG for Bladder Cancer. Cells, 9(8), 1839. https://doi.org/10.3390/cells9081839