Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6




Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Isolation and Generation of hLECs
2.3. Quantitation of Intracellular ROS Level by 2’-7’-Dichlorofluorescein Diacetate (H2-DCF-DA) and CellROX® Deep Red Reagent
2.4. Real-Time Quantitative Reverse Transcriptase-Polymerase Chain Reaction (RT-qPCR)
2.5. Eukaryotic Plasmids
2.6. Construction of Prdx6 Antisense (Prdx6-As)
2.7. Lentiviral (LV) Infection
2.8. Western Blotting
2.9. Chromatin Immunoprecipitation (ChIP)-qPCR Assay
2.10. Construction of Human Prdx6 Promoter-Chloramphenicol Acetyltransferase (CAT) Reporter Vector
2.11. Site-Directed Mutagenesis (SDM)
2.11.1. Bmal1/E-Box SDM Primer:
2.11.2. Nrf2/ARE SDM Primer:
2.12. Cell Survival Assay (MTS Assay)
2.13. Animal Studies for Zeitgeber Time (ZT)
2.13.1. Collection of Lenses and mRNA analysis
2.13.2. Collection of Lenses and Protein Isolation
2.13.3. Quantitation of Intracellular ROS Level by H2-DCF-DA in Mouse Eye Lens Ex-Vivo
2.14. Statistical Analysis
3. Results
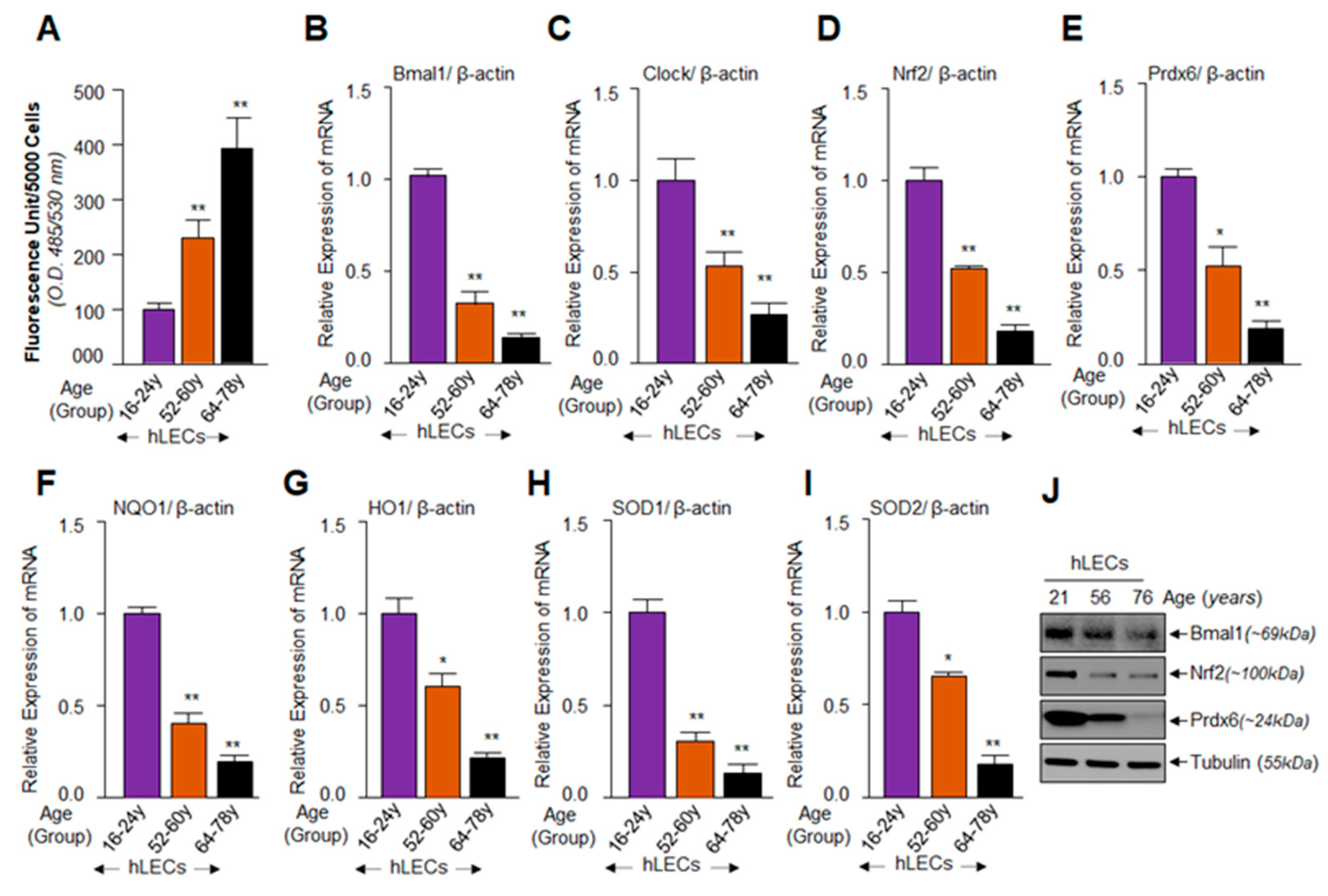
3.1. Increased ROS Levels with Advancing Age Were Associated with a Progressive Decline of Clock Gene Bmal1-Clock, and Nrf2 and Nrf2/ARE-Dependent Antioxidant Enzymes
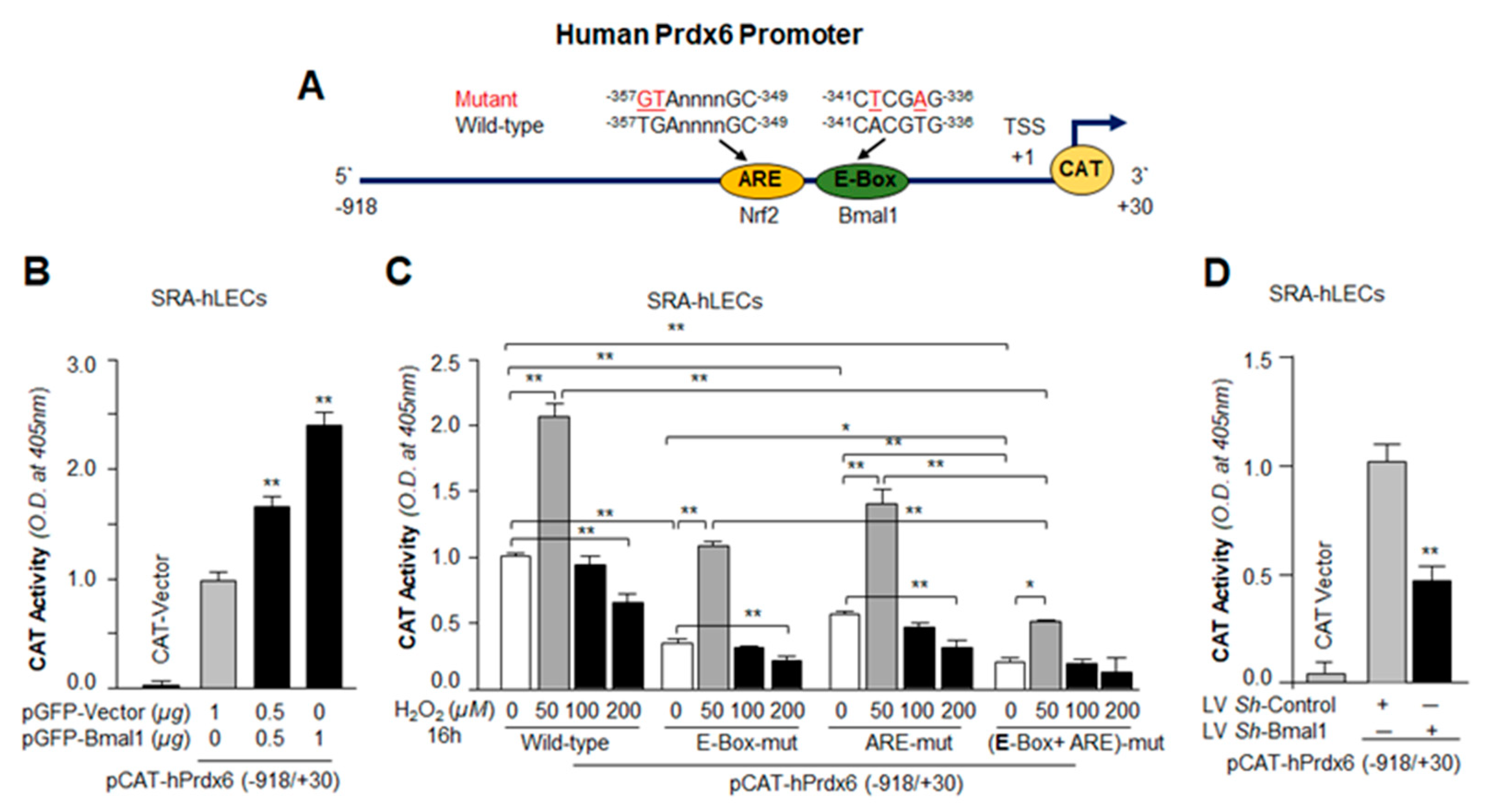
3.2. Bmal1-Overexpression Augmented Expression of Nrf2 and Nrf2/ARE–Dependent Antioxidants in a Dose-Dependent Fashion
3.3. Bmal1-Deficient SRA-hLECs Showed Down-Regulation of Nrf2/ARE Pathway as Observed in Aging Cells
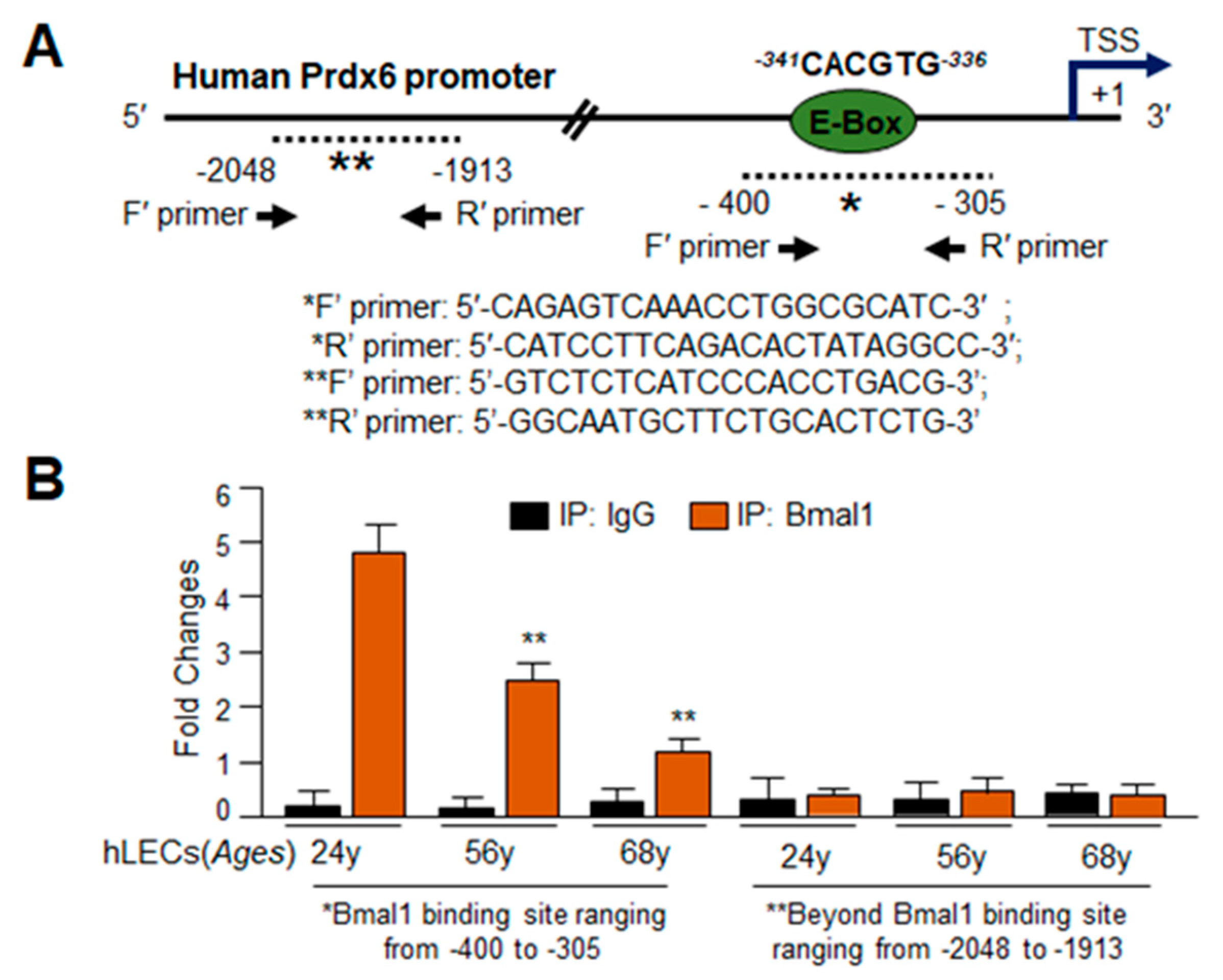
3.4. In Silico Analyses and DNA-Binding Assay Disclosed Presence of Active Bmal1/E-Box Responsive Element in Prdx6 Gene Promoter, Which Was Functionally Dysregulated in Aging
3.5. In Vivo DNA Binding Assay Showed That Bmal1 Enrichment at E-Box Sequences in the Prdx6 Promoter Was Dependent on Its Cellular Abundance
3.6. Transactivation Analysis Disclosed that Bmal1 and Nrf2 Cooperatively Regulated Prdx6 Transcription
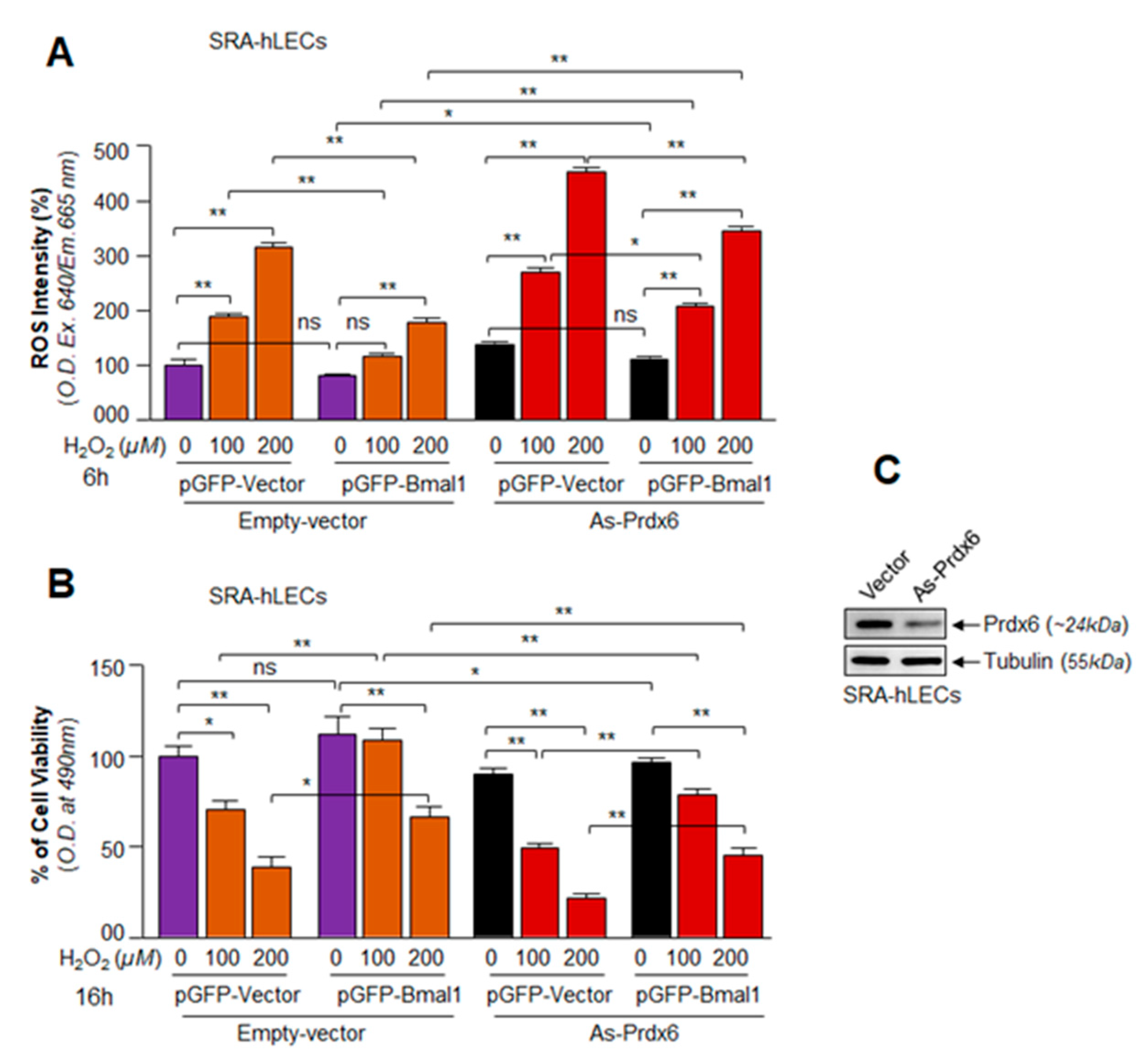
3.7. A Cellular Abundance of Prdx6 Was Required for a Significant Protection of hLECs by Bmal1 against Oxidative Stress
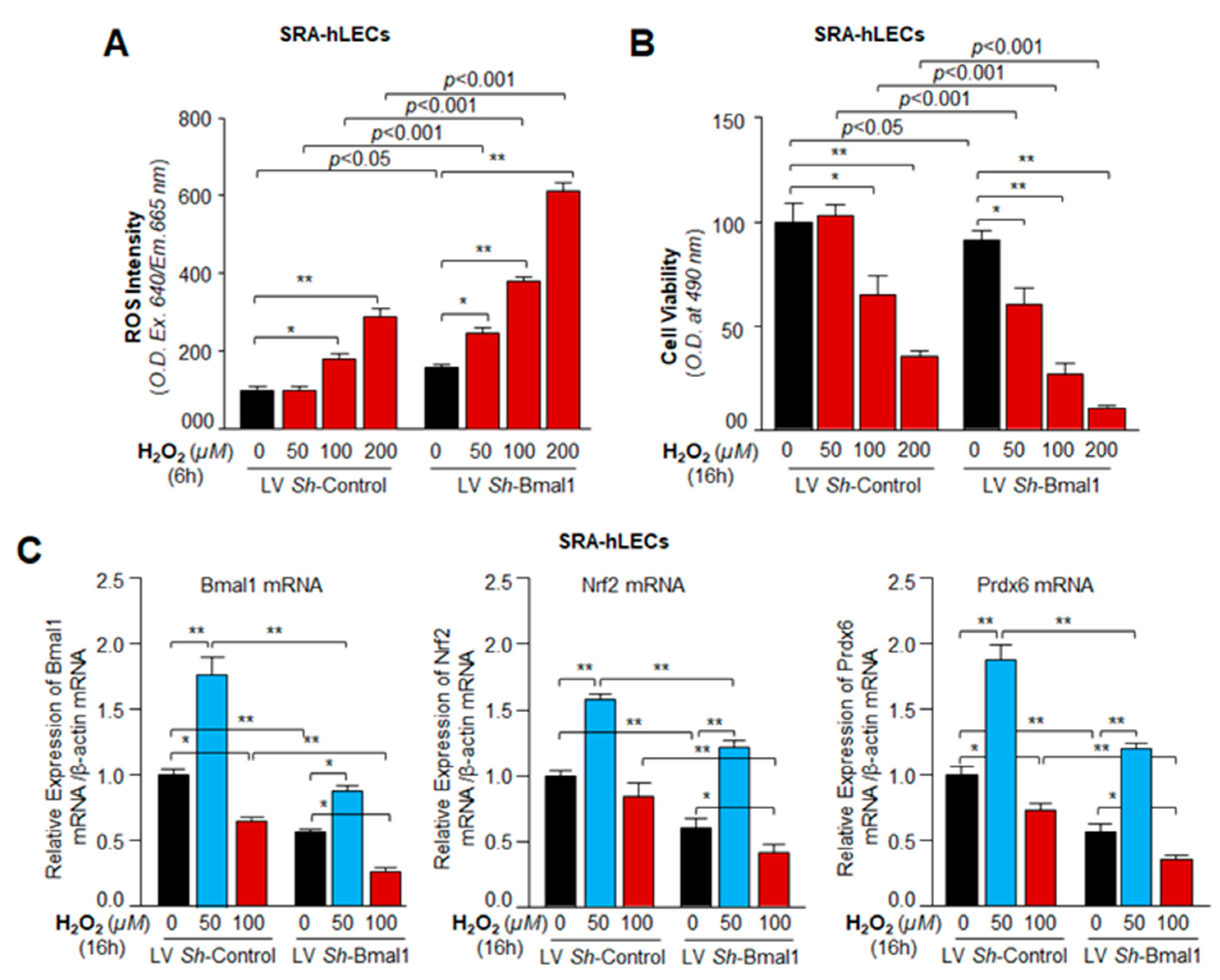
3.8. Bmal1 Knockdown Showed That Bmal1 Expression in LECs Was Required for Cellular Resistance against Oxidative Stress through Nrf2-Driven Antioxidant Pathway
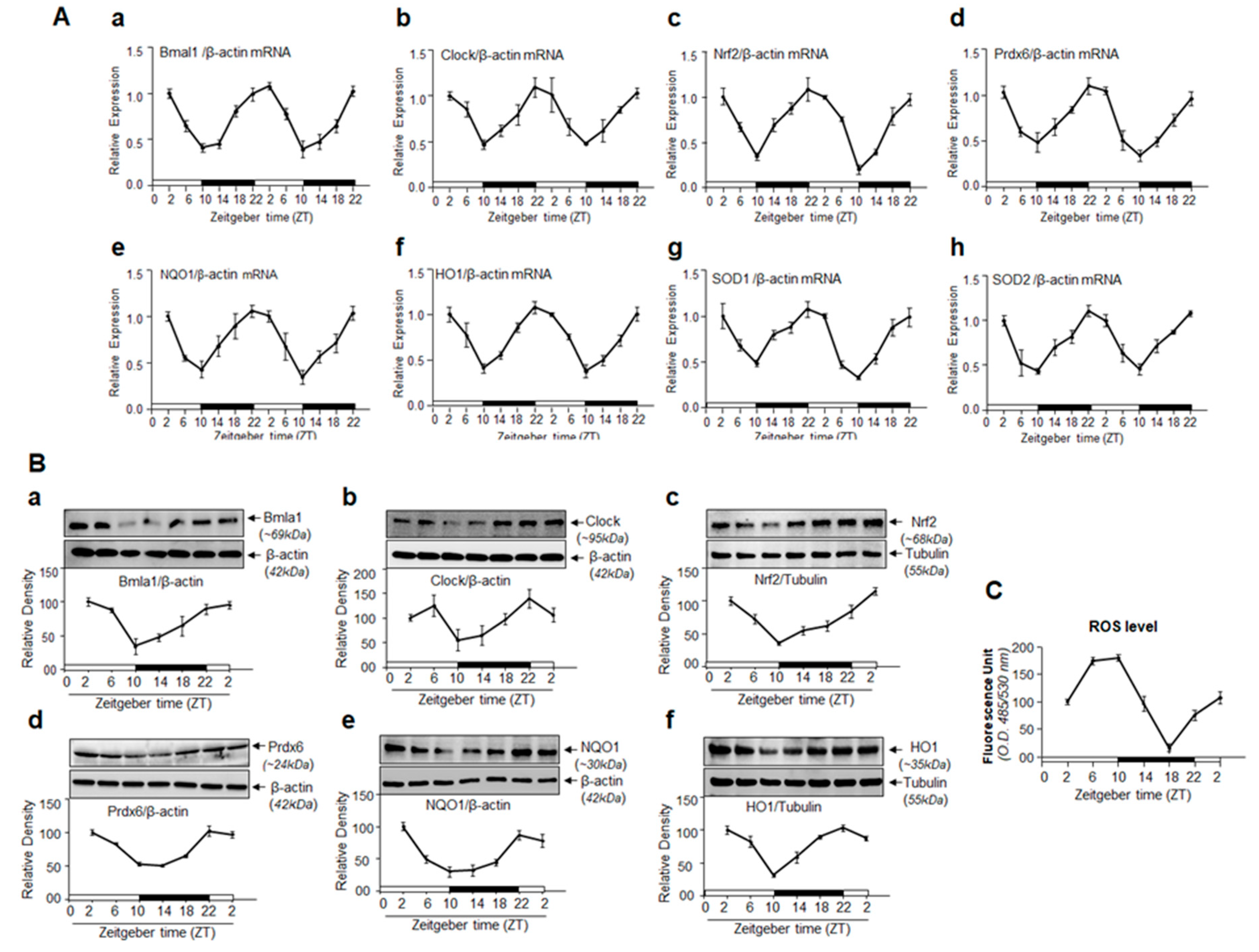
3.9. Circadian Expression Profiles of Core Clock Genes, Nrf2 and Nrf2-Dependent Phase II Antioxidant Genes Are Reciprocally Associated with ROS Levels in C57BL/6 Mouse Eye Lens
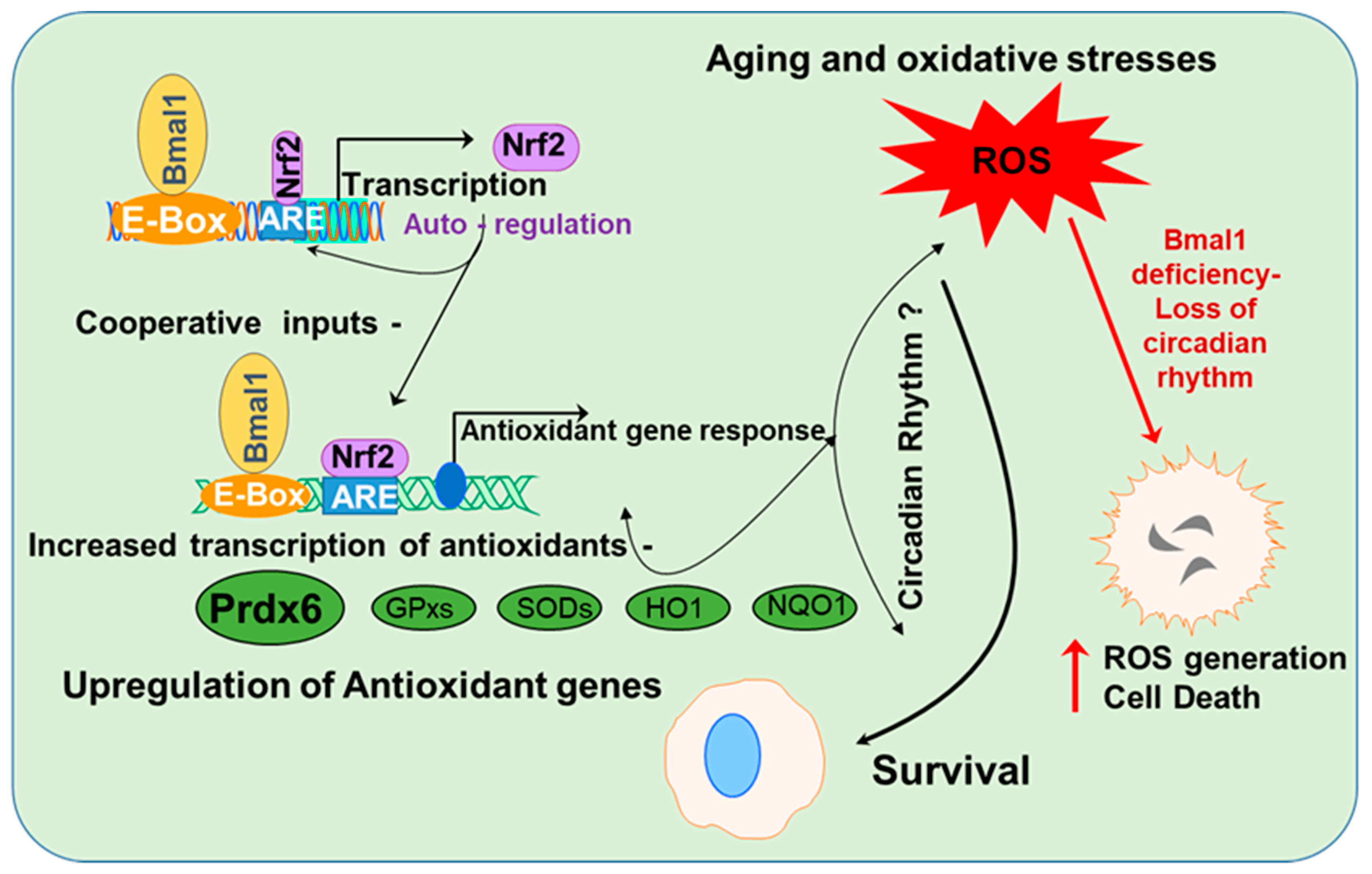
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khapre, R.V.; Kondratova, A.A.; Susova, O.; Kondratov, R.V. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 2011, 10, 4162–4169. [Google Scholar] [CrossRef] [Green Version]
- Gachon, F.; Bonnefont, X. Circadian clock-coordinated hepatic lipid metabolism: Only transcriptional regulation? Aging (Albany Ny) 2010, 2, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Dubrovsky, Y.V.; Samsa, W.E.; Kondratov, R.V. Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging (Albany Ny) 2010, 2, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Wible, R.S.; Ramanathan, C.; Sutter, C.H.; Olesen, K.M.; Kensler, T.W.; Liu, A.C.; Sutter, T.R. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. Elife 2018, 7, e31656. [Google Scholar] [CrossRef] [Green Version]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekovic-Vaughan, V.; Gibbs, J.; Yoshitane, H.; Yang, N.; Pathiranage, D.; Guo, B.; Sagami, A.; Taguchi, K.; Bechtold, D.; Loudon, A.; et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014, 28, 548–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Welz, P.S.; Zinna, V.M.; Symeonidi, A.; Koronowski, K.B.; Kinouchi, K.; Smith, J.G.; Guillen, I.M.; Castellanos, A.; Furrow, S.; Aragon, F.; et al. BMAL1-Driven Tissue Clocks Respond Independently to Light to Maintain Homeostasis. Cell 2019, 177, 1436–1447. [Google Scholar] [CrossRef]
- Hood, S.; Amir, S. The aging clock: Circadian rhythms and later life. J. Clin. Investig. 2017, 127, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Musiek, E.S.; Hu, K.; Cappuccio, F.P.; Yaffe, K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019, 18, 307–318. [Google Scholar] [CrossRef]
- Storch, K.F.; Lipan, O.; Leykin, I.; Viswanathan, N.; Davis, F.C.; Wong, W.H.; Weitz, C.J. Extensive and divergent circadian gene expression in liver and heart. Nature 2002, 417, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.B.; Karp, N.A.; Maywood, E.S.; Sage, E.A.; Deery, M.; O’Neill, J.S.; Wong, G.K.; Chesham, J.; Odell, M.; Lilley, K.S.; et al. Circadian orchestration of the hepatic proteome. Curr. Biol. 2006, 16, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Lamia, K.A.; Storch, K.F.; Weitz, C.J. Physiological significance of a peripheral tissue circadian clock. Proc. Natl. Acad. Sci. USA 2008, 105, 15172–15177. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Stangherlin, A.; Reddy, A.B. Regulation of circadian clocks by redox homeostasis. J. Biol. Chem. 2013, 288, 26505–26511. [Google Scholar] [CrossRef] [Green Version]
- Kondratov, R.V.; Vykhovanets, O.; Kondratova, A.A.; Antoch, M.P. Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging (Albany Ny) 2009, 1, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Wakabayashi, N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry 2005, 44, 6889–6899. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharm. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shibata, T.; Takaya, K.; Shiraishi, K.; Kohno, T.; Kunitoh, H.; Tsuta, K.; Furuta, K.; Goto, K.; Hosoda, F.; et al. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol. Cell. Biol. 2013, 33, 2402–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ma, Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharm. 2009, 76, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Dodia, C.; Chatterjee, S.; Zagorski, J.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Jain, M.; Fisher, A.B. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. J. Pharm. Exp. 2013, 345, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.Q.; Zhang, D.; Jin, T.; Cai, D.J.; Wu, Q.; Lu, Y.; Liu, J.; Klaassen, C.D. Diurnal variation of hepatic antioxidant gene expression in mice. PLoS ONE 2012, 7, e44237. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ci, X.; Lv, H.; Wang, L.; Wang, X.; Peng, L.; Qin, F.X.; Cheng, G. The antioxidative potential of farrerol occurs via the activation of Nrf2 mediated HO-1 signaling in RAW 264.7 cells. Chem. Biol. Interact. 2015, 239, 192–199. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Hasanova, N.; Tanaka, Y.; Fatma, N.; Takamura, Y.; Singh, D.P.; Akagi, Y. Protein expression profiling of lens epithelial cells from Prdx6-depleted mice and their vulnerability to UV radiation exposure. Am. J. Physiol. Cell Physiol. 2010, 298, C342–C354. [Google Scholar] [CrossRef]
- Chhunchha, B.; Fatma, N.; Bhargavan, B.; Kubo, E.; Kumar, A.; Singh, D.P. Specificity protein, Sp1-mediated increased expression of Prdx6 as a curcumin-induced antioxidant defense in lens epithelial cells against oxidative stress. Cell Death Dis. 2011, 2, e234. [Google Scholar] [CrossRef]
- Chhunchha, B.; Singh, P.; Stamer, W.D.; Singh, D.P. Prdx6 retards senescence and restores trabecular meshwork cell health by regulating reactive oxygen species. Cell Death Discov. 2017, 3, 17060. [Google Scholar] [CrossRef] [Green Version]
- Chhunchha, B.; Kubo, E.; Singh, P.; Singh, D.P. Sumoylation-deficient Prdx6 repairs aberrant Sumoylation-mediated Sp1 dysregulation-dependent Prdx6 repression and cell injury in aging and oxidative stress. Aging (Albany Ny) 2018, 10, 2284–2315. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, S.P.; Singh, D.P. Delivery of a protein transduction domain-mediated Prdx6 protein ameliorates oxidative stress-induced injury in human and mouse neuronal cells. Am. J. Physiol. Cell Physiol. 2016, 310, C1–C16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Rai, P.; Singh, S.P.; Singh, D.P. Curcumin abates hypoxia-induced oxidative stress based-ER stress-mediated cell death in mouse hippocampal cells (HT22) by controlling Prdx6 and NF-kappaB regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C636–C655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, D.P. Aberrant sumoylation signaling evoked by reactive oxygen species impairs protective function of Prdx6 by destabilization and repression of its transcription. FEBS J. 2014, 281, 3357–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.P.; Bhargavan, B.; Chhunchha, B.; Kubo, E.; Kumar, A.; Fatma, N. Transcriptional protein Sp1 regulates LEDGF transcription by directly interacting with its cis-elements in GC-rich region of TATA-less gene promoter. PLoS ONE 2012, 7, e37012. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Moulik, M.; Fang, Z.; Saha, P.; Zou, F.; Xu, Y.; Nelson, D.L.; Ma, K.; Moore, D.D.; Yechoor, V.K. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol. Cell. Biol. 2013, 33, 2327–2338. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Li, L.; Wang, J.; Zhao, B.; Pan, J.; Wang, L.; Liu, X.; Liu, X.; Liu, Z. Resveratrol Prevents Acrylamide-Induced Mitochondrial Dysfunction and Inflammatory Responses via Targeting Circadian Regulator Bmal1 and Cry1 in Hepatocytes. J. Agric. Food Chem. 2019, 67, 8510–8519. [Google Scholar] [CrossRef]
- Qi, G.; Wu, W.; Mi, Y.; Shi, R.; Sun, K.; Li, R.; Liu, X.; Liu, X. Tea polyphenols direct Bmal1-driven ameliorating of the redox imbalance and mitochondrial dysfunction in hepatocytes. Food Chem. Toxicol. 2018, 122, 181–193. [Google Scholar] [CrossRef]
- Albrecht, U. Timing to perfection: The biology of central and peripheral circadian clocks. Neuron 2012, 74, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Ibaraki, N.; Chen, S.C.; Lin, L.R.; Okamoto, H.; Pipas, J.M.; Reddy, V.N. Human lens epithelial cell line. Exp. Eye Res. 1998, 67, 577–585. [Google Scholar] [CrossRef]
- Singh, D.P.; Kubo, E.; Takamura, Y.; Shinohara, T.; Kumar, A.; Chylack, L.T.; Fatma, N. DNA binding domains and nuclear localization signal of LEDGF: Contribution of two helix-turn-helix (HTH)-like domains and a stretch of 58 amino acids of the N-terminal to the trans-activation potential of LEDGF. J. Mol. Biol. 2006, 355, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Kubo, E.; Fatma, N.; Akagi, Y.; Beier, D.R.; Singh, S.P.; Singh, D.P. TAT-mediated PRDX6 protein transduction protects against eye lens epithelial cell death and delays lens opacity. Am. J. Physiol. Cell Physiol. 2008, 294, C842–C855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatma, N.; Kubo, E.; Sharma, P.; Beier, D.R.; Singh, D.P. Impaired homeostasis and phenotypic abnormalities in Prdx6-/-mice lens epithelial cells by reactive oxygen species: Increased expression and activation of TGFbeta. Cell Death Differ. 2005, 12, 734–750. [Google Scholar] [CrossRef] [PubMed]
- McAvoy, J.; Chamberlain, C.; de Longh, R.; Hales, A.; Lovicu, F. Lens development. Eye 1999, 13, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatigorsky, J.; Rothschild, S.S. Loss during development of the ability of chick embryonic lens cells to elongate in culture: Inverse relationship between cell division and elongation. Dev. Biol. 1972, 28, 382–389. [Google Scholar] [CrossRef]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar] [CrossRef]
- Fatma, N.; Singh, P.; Chhunchha, B.; Kubo, E.; Shinohara, T.; Bhargavan, B.; Singh, D.P. Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C954–C967. [Google Scholar] [CrossRef]
- Chhunchha, B.; Singh, P.; Singh, D.P.; Kubo, E. Ginkgolic Acid Rescues Lens Epithelial Cells from Injury Caused by Redox Regulated-Aberrant Sumoylation Signaling by Reviving Prdx6 and Sp1 Expression and Activities. Int. J. Mol. Sci. 2018, 19, 3520. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Miyazawa, T.; Fatma, N.; Akagi, Y.; Singh, D.P. Development- and age-associated expression pattern of peroxiredoxin 6, and its regulation in murine ocular lens. Mech. Ageing Dev. 2006, 127, 249–256. [Google Scholar] [CrossRef]
- Kubo, E.; Singh, D.P.; Fatma, N.; Akagi, Y. TAT-mediated peroxiredoxin 5 and 6 protein transduction protects against high-glucose-induced cytotoxicity in retinal pericytes. Life Sci. 2009, 84, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Naito, H.; Takahashi, R.; Jung, K.J.; Kim, H.J.; Goto, S. Age-associated increase in oxidative stress and nuclear factor kappaB activation are attenuated in rat liver by regular exercise. FASEB J. 2004, 18, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.; Gustafsson Trajkovska, M.; Perman-Sundelin, J.; Fogelstrand, P.; Adiels, M.; Johansson, M.; Mattsson-Hulten, L.; Boren, J.; Levin, M. Imaging of Intracellular and Extracellular ROS Levels in Atherosclerotic Mouse Aortas Ex Vivo: Effects of Lipid Lowering by Diet or Atorvastatin. PLoS ONE 2015, 10, e0130898. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Velingkaar, N.S.; Kondratov, R.V. Transcriptional control of antioxidant defense by the circadian clock. Antioxid. Redox Signal. 2014, 20, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Tamaru, T.; Hattori, M.; Ninomiya, Y.; Kawamura, G.; Vares, G.; Honda, K.; Mishra, D.P.; Wang, B.; Benjamin, I.; Sassone-Corsi, P.; et al. ROS stress resets circadian clocks to coordinate pro-survival signals. PLoS ONE 2013, 8, e82006. [Google Scholar] [CrossRef] [Green Version]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Sussman, W.; Stevenson, M.; Mowdawalla, C.; Mota, S.; Ragolia, L.; Pan, X. BMAL1 controls glucose uptake through paired-homeodomain transcription factor 4 in differentiated Caco-2 cells. Am. J. Physiol. Cell Physiol. 2019, 317, C492–C501. [Google Scholar] [CrossRef]
- Ji, G.; Lv, K.; Chen, H.; Wang, Y.; Zhang, Y.; Li, Y.; Qu, L. Hydrogen peroxide modulates clock gene expression via PRX2-STAT3-REV-ERBalpha/beta pathway. Free Radic. Biol. Med. 2019, 145, 312–320. [Google Scholar] [CrossRef]
- Lee, J.; Ma, K.; Moulik, M.; Yechoor, V. Untimely oxidative stress in beta-cells leads to diabetes-Role of circadian clock in beta-cell function. Free Radic. Biol. Med. 2018, 119, 69–74. [Google Scholar] [CrossRef]
- Bunger, M.K.; Wilsbacher, L.D.; Moran, S.M.; Clendenin, C.; Radcliffe, L.A.; Hogenesch, J.B.; Simon, M.C.; Takahashi, J.S.; Bradfield, C.A. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 2000, 103, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Yue, J.; Zhang, Y.; Zhou, L.; Hao, W.; Yuan, J.; Qiang, B.; Ding, J.M.; Peng, X.; Cao, J.M. CLOCK/BMAL1 regulates human nocturnin transcription through binding to the E-box of nocturnin promoter. Mol. Cell. Biochem. 2008, 317, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Bokov, A.; Chaudhuri, A.; Richardson, A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004, 125, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Masoro, E.J.; Nelson, J.F.; Strong, R.; McMahan, C.A.; Richardson, A. Genetic mouse models of extended lifespan. Exp. Gerontol. 2003, 38, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Pinazo-Duran, M.D.; Gallego-Pinazo, R.; Garcia-Medina, J.J.; Zanon-Moreno, V.; Nucci, C.; Dolz-Marco, R.; Martinez-Castillo, S.; Galbis-Estrada, C.; Marco-Ramirez, C.; Lopez-Galvez, M.I.; et al. Oxidative stress and its downstream signaling in aging eyes. Clin. Interv. Aging 2014, 9, 637–652. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Sussan, T.E.; Gajghate, S.; Chatterjee, S.; Mandke, P.; McCormick, S.; Sudini, K.; Kumar, S.; Breysse, P.N.; Diette, G.B.; Sidhaye, V.K.; et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L27–L36. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, I.; Fisher, A.B.; Christofidou-Solomidou, M.; Gao, L.; Tao, J.Q.; Sorokina, E.M.; Lien, Y.C.; Bates, S.R.; Feinstein, S.I. Keratinocyte growth factor and glucocorticoid induction of human peroxiredoxin 6 gene expression occur by independent mechanisms that are synergistic. Antioxid. Redox Signal. 2014, 20, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor, F.F., 3rd; Miller, B.F.; Hamilton, K.L. Nrf2 Signaling and the Slowed Aging Phenotype: Evidence from Long-Lived Models. Oxid. Med. Cell Longev. 2015, 2015, 732596. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manevich, Y.; Shuvaeva, T.; Dodia, C.; Kazi, A.; Feinstein, S.I.; Fisher, A.B. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A(2) activities. Arch. Biochem. Biophys. 2009, 485, 139–149. [Google Scholar] [CrossRef] [Green Version]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Antoch, M.P.; Kondratov, R.V. Circadian proteins and genotoxic stress response. Circ. Res. 2010, 106, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.F.; Li, X.K.; Li, W.Q.; Gao, Q.; Zhang, Y.; Wang, X.M.; Fu, J.Q.; Cui, S.S.; Qu, J.H.; Zhao, X.; et al. Diurnal oscillations of endogenous H2O2 sustained by p66(Shc) regulate circadian clocks. Nat. Cell Biol. 2019, 21, 1553–1564. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, D.; Liu, W.; Li, S.; Chen, J.; Shen, Y.; Wang, F.; Hu, L.F.; Liu, C.F. Disruption of the Circadian Clock Alters Antioxidative Defense via the SIRT1-BMAL1 Pathway in 6-OHDA-Induced Models of Parkinson’s Disease. Oxid. Med. Cell. Longev. 2018, 2018, 4854732. [Google Scholar] [CrossRef]
- Kuruvilla, K.P.; Nandhu, M.S.; Paul, J.; Paulose, C.S. Oxidative stress mediated neuronal damage in the corpus striatum of 6-hydroxydopamine lesioned Parkinson’s rats: Neuroprotection by serotonin, GABA and bone marrow cells supplementation. J. Neurol. Sci. 2013, 331, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Buhr, E.D.; Yoo, S.H.; Takahashi, J.S. Temperature as a universal resetting cue for mammalian circadian oscillators. Science 2010, 330, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagoshi, E.; Saini, C.; Bauer, C.; Laroche, T.; Naef, F.; Schibler, U. Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 2004, 119, 693–705. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | RT-qPCR Forward Primers | RT-qPCR Reverse Primers |
|---|---|---|
| hBmal1 | 5′-GGAAAAATAGGCCGAATGAT-3′ | 5′-TGAGCCTGGCCTGATAGTAG-3′ |
| hClock | 5′-GAGAGCGCGAAGGAAATCT-3′ | 5′-AGCAGCTTTGCAGGAACAA-3′ |
| hNrf2 | 5′-TGCTTTATAGCGTGCAAACCTCGC-3′ | 5′-ATCCATGTCCCTTGACAGCACAGA-3′ |
| hPrdx6 | 5′-GCATCCGTTTCCACGACT-3′ | 5′-TGCACACTGGGGTAAAGTCC-3′ |
| hNQO1 | 5′-ATGTATGACAAAGGACCCTTCC-3′ | 5′-TCCCTTGCAGAGAGTACATGG-3′ |
| hHO1 | 5′-GGCAGAGGGTGATAGAAGAGG-3′ | 5′-AGCTCCTGCAACTCCTCAAA-3′ |
| hSOD1 | 5′-TCATCAATTTCGAGCAGAAGG-3′ | 5′-CAGGCCTTCAGTCAGTCCTTT-3′ |
| hSOD2 | 5′-AAGTACCAGGAGGCGTTGG-3′ | 5′-TGAACTTCAGTGCAGGCTGA-3′ |
| hβ-actin | 5′-CCAACCGCGAGAAGATGA-3′ | 5′-CCAGAGGCGTACAGGGATAG-3′ |
| Gene | RT-qPCR Forward Primer | RT-qPCR Reverse Primer |
|---|---|---|
| mBmal1 | 5′-TTTGGGCTAGCTGTGGATAG-3′ | 5′-AAATATCCACATGGGGGACT-3′ |
| mClock | 5′-CAGCTTCCTTCAGTTCAGCA-3′ | 5′-CCGTGGAGCAACCTAGATGT-3′ |
| mNrf2 | 5′-TCTCCTCGCTGGAAAAAGAA-3′ | 5′-AATGTGCTGGCTGTGCTTTA-3′ |
| mPrdx6 | 5′-TTCAATAGACAGTGTTGAGGATCA-3′ | 5′-CGTGGGTGTTTCACCATTG-3′ |
| mNQO1 | 5′-AGCGTTCGGTATTACGATCC-3′ | 5′-AGTACAATCAGGGCTCTTCTCG-3′ |
| mHO1 | 5′-AGGCTAAGACCGCCTTCCT-3′ | 5′-TGTGTTCCTCTGTCAGCATCA-3′ |
| mSOD1 | 5′-CAGGACCTCATTTTAATCCTCAC-3′ | 5′-TGCCCAGGTCTCCAACAT-3′ |
| mSOD2 | 5′-TGCTCTAATCAGGACCCATTG-3′ | 5′-GTAGTAAGCGTGCTCCCACAC-3′ |
| mβ-actin | 5′-CTAAGGCCAACCGTGAAAAG-3′ | 5′-ACCAGAGGCATACAGGGACA-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhunchha, B.; Kubo, E.; Singh, D.P. Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells 2020, 9, 1861. https://doi.org/10.3390/cells9081861
Chhunchha B, Kubo E, Singh DP. Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells. 2020; 9(8):1861. https://doi.org/10.3390/cells9081861
Chicago/Turabian StyleChhunchha, Bhavana, Eri Kubo, and Dhirendra P. Singh. 2020. "Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6" Cells 9, no. 8: 1861. https://doi.org/10.3390/cells9081861
APA StyleChhunchha, B., Kubo, E., & Singh, D. P. (2020). Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells, 9(8), 1861. https://doi.org/10.3390/cells9081861