Structural and Mechanical Aberrations of the Nuclear Lamina in Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structural Support Provided by Nuclear Lamins
3. Role of Lamins in Mechanosensing
4. The Nuclear Lamina in Laminopathies
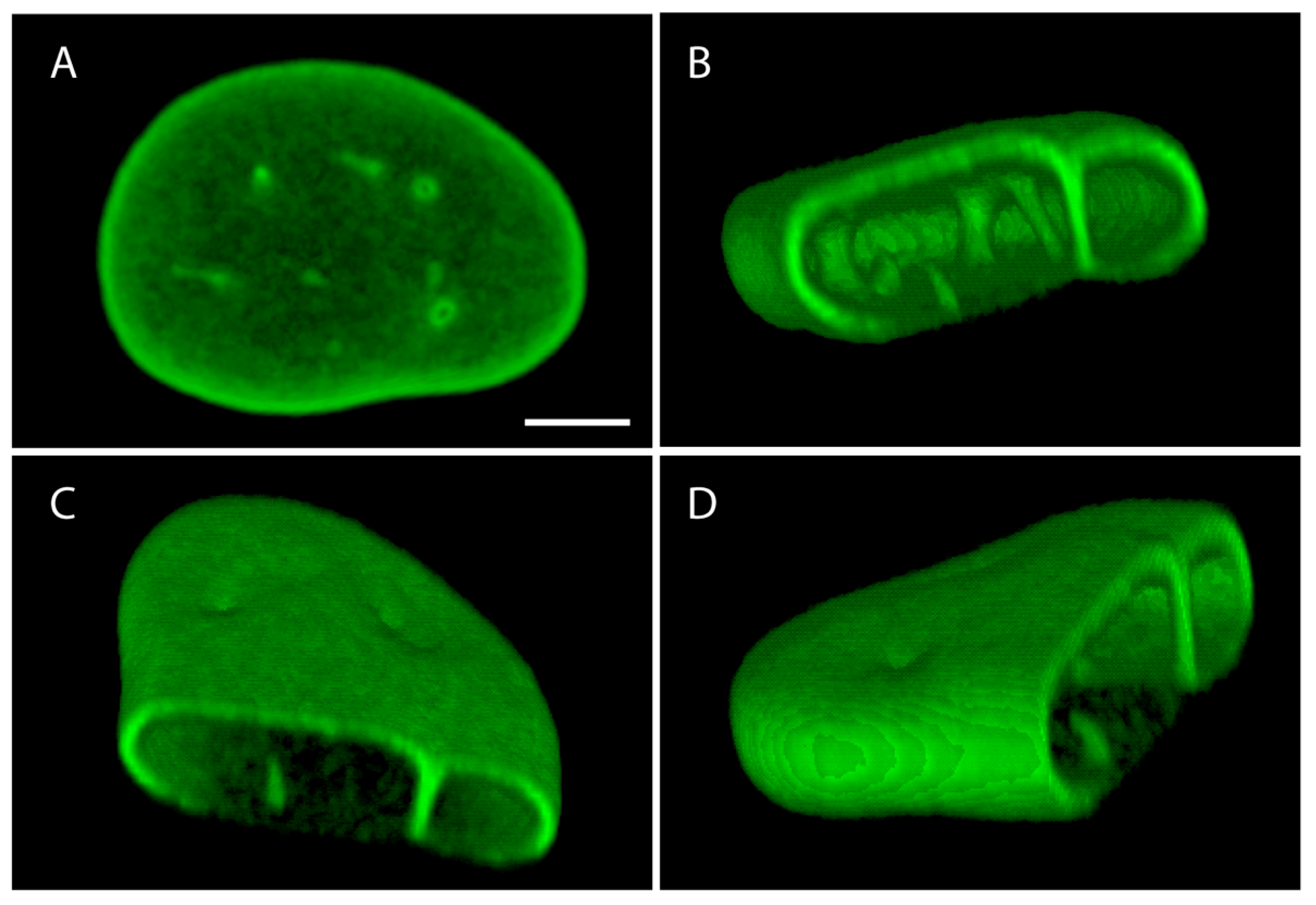
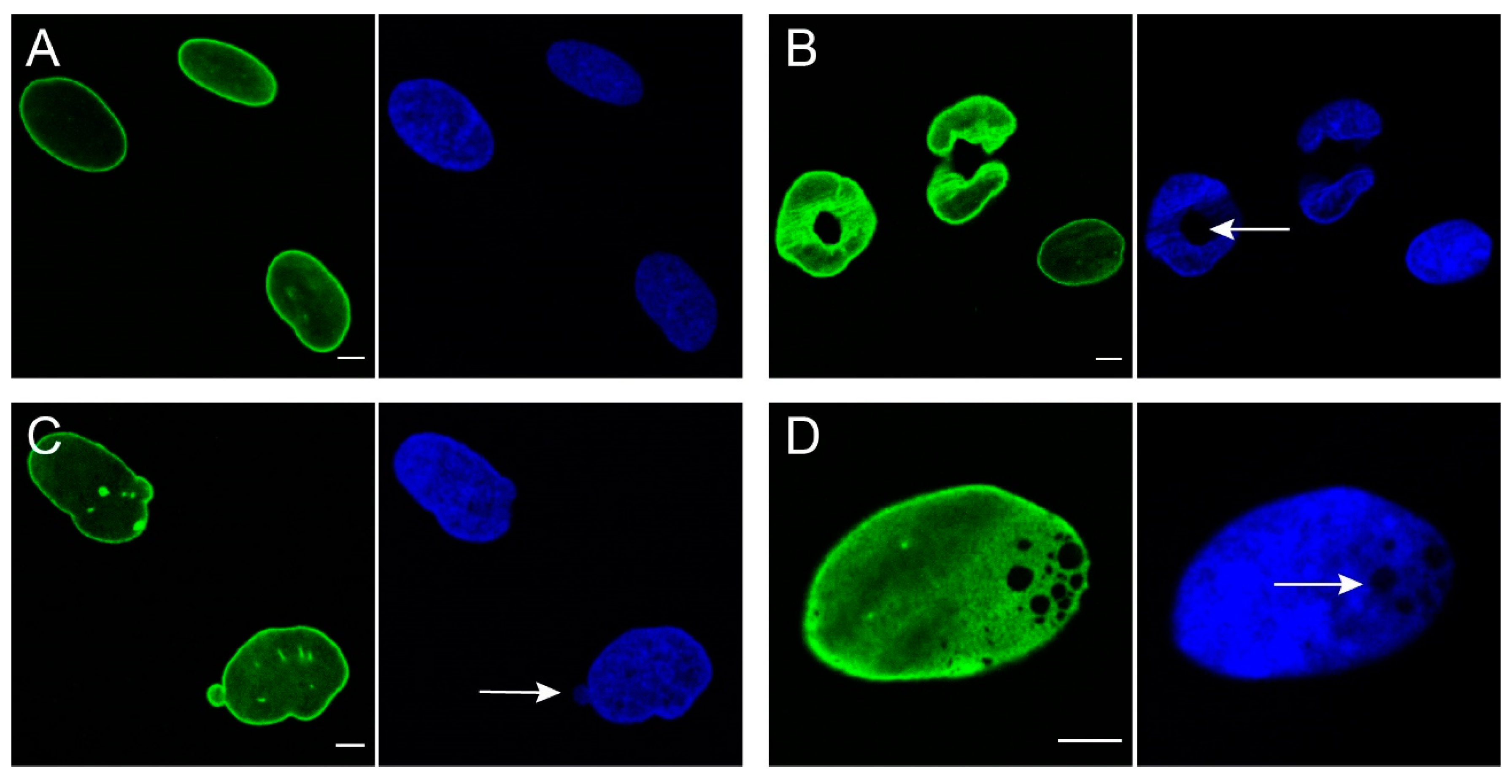
4.1. Changes in Nuclear Morphology
4.2. Changes in Structural and Mechanical Properties
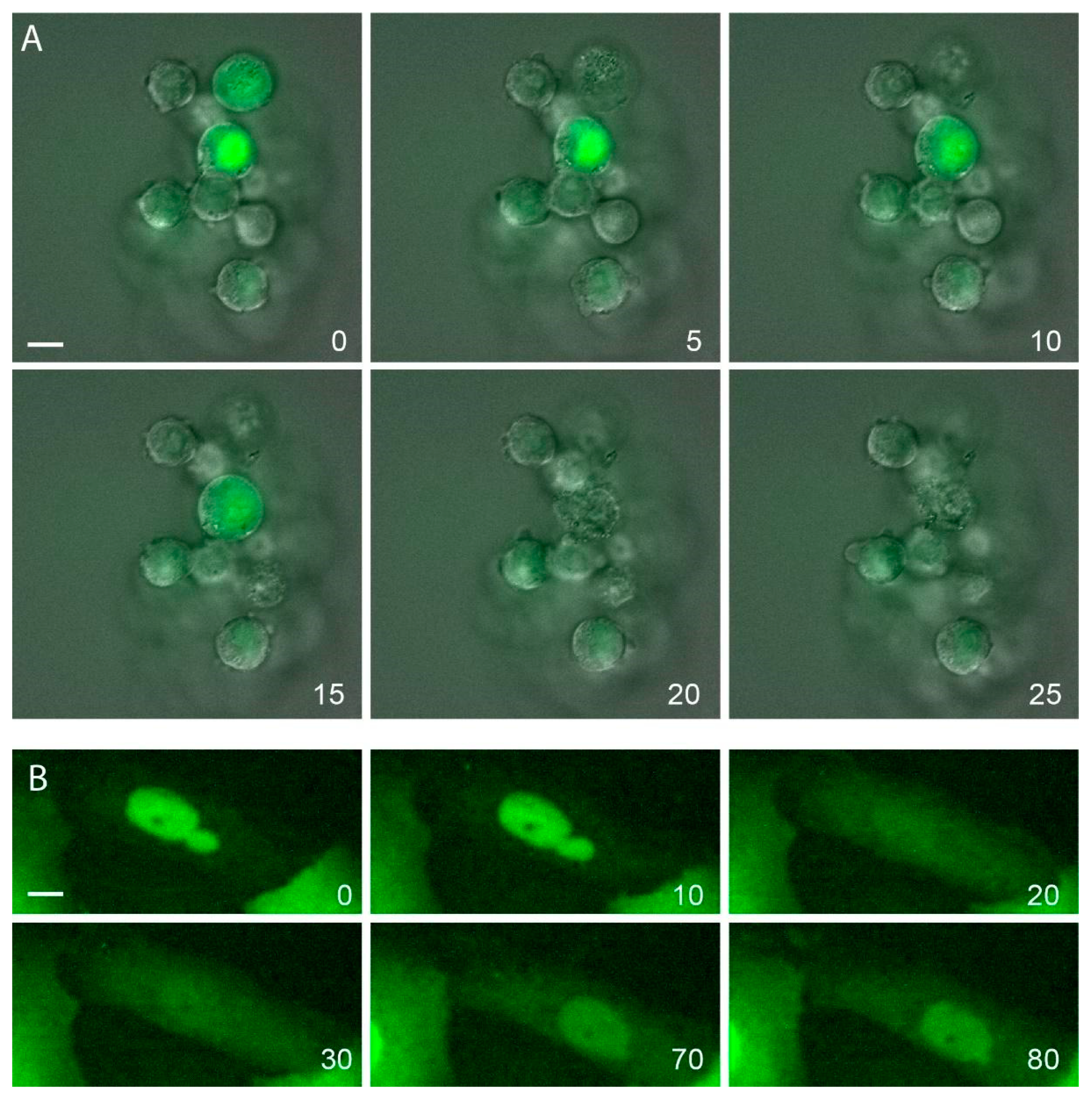
4.2.1. Nuclear Envelope Rupture
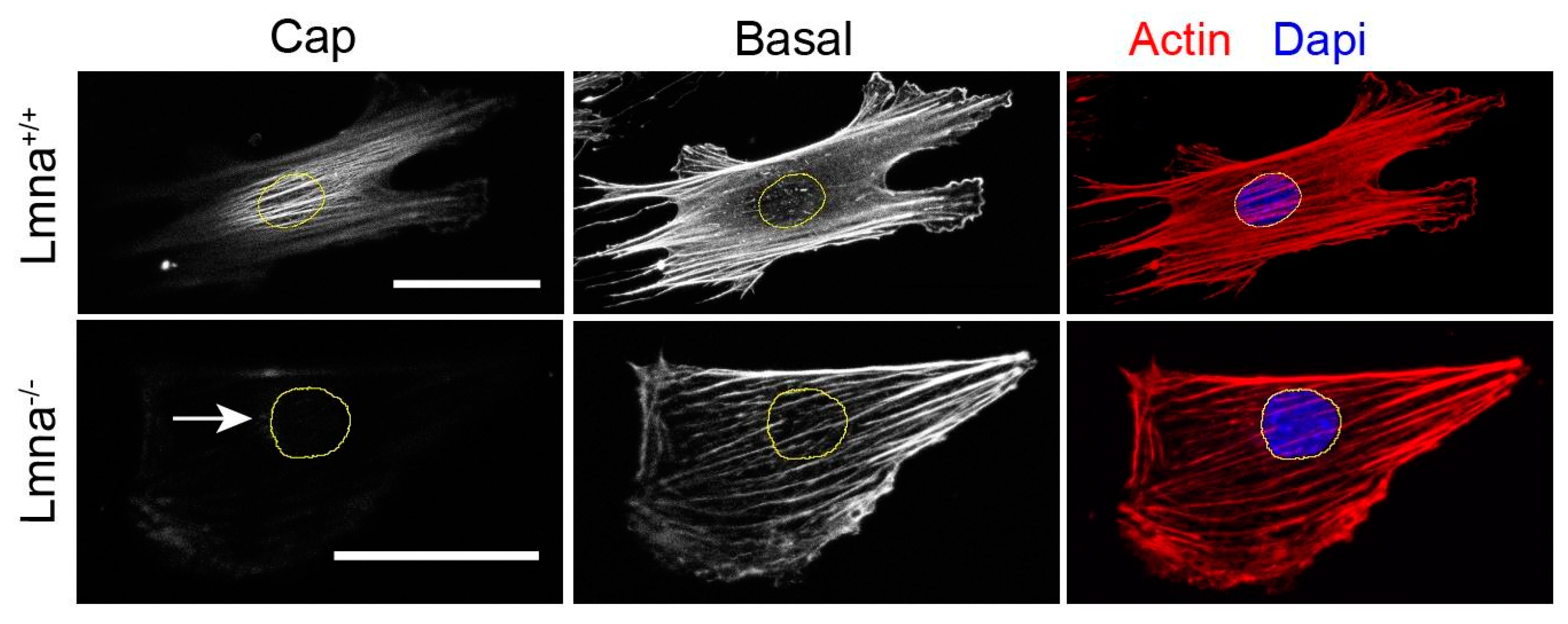
4.2.2. Actin Cytoskeletal Organization Defects
4.2.3. Signaling Pathway Alterations
4.2.4. Structural Defects Explaining Laminopathy Symptoms
5. The Nuclear Lamina in Cancer
6. The Nuclear Lamina in Viral Infections
6.1. Herpesviruses
6.2. Human Polyomaviruses
6.3. Baculoviruses
6.4. Parvoviruses
6.5. Retroviruses
7. Conclusions and Recommendations
Funding
Acknowledgments
Conflicts of Interest
References
- Gruenbaum, Y.; Margalit, A.; Goldman, R.D.; Shumaker, D.K.; Wilson, K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.V.; Ramaekers, F.C.S.; Bonne, G.; Ben Yaou, R.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, N.; Heins, S.; Aebi, U. Nuclear lamins: Their structure, assembly, and interactions. J. Struct. Biol. 1998, 122, 42–66. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Goldman, R.D.; Meyuhas, R.; Mills, E.; Margalit, A.; Fridkin, A.; Dayani, Y.; Prokocimer, M.; Enosh, A. The nuclear lamina and its functions in the nucleus. Int. Rev. Cytol. 2003, 226, 1–62. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear lamins: Thin filaments with major functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Medalia, O. Lamins: The structure and protein complexes. Curr. Opin. Cell Biol. 2015, 32, 7–12. [Google Scholar] [CrossRef]
- Tatli, M.; Medalia, O. Insight into the functional organization of nuclear lamins in health and disease. Curr. Opin. Cell Biol. 2018, 54, 72–79. [Google Scholar] [CrossRef]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef]
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Aebi, U.; Cohn, J.; Buhle, L.; Gerace, L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature 1986, 323, 560–564. [Google Scholar] [CrossRef]
- Turgay, Y.; Medalia, O. The structure of lamin filaments in somatic cells as revealed by cryo-electron tomography. Nucleus 2017, 8, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, T.; Maass, K.; Hergt, M.; Aebi, U.; Herrmann, H. Lamin A and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus 2011, 2, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Gesson, K.; Foisner, R. Lamina-independent lamins in the nuclear interior serve important functions. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Chojnowski, A.; Boudier, T.; Ser, Z.; Stewart, C.; Burke, B. A-type lamins form distinct filamentous networks with differential nuclear pore complex associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef] [Green Version]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A-and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.; Stepen, A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4085. [Google Scholar] [CrossRef]
- Broers, J.L.V.; Kuijpers, H.J.H.; Östlund, C.; Worman, H.J.; Endert, J.; Ramaekers, F.C.S. Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp. Cell Res. 2005, 304, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Drozdz, M.M.; Jiang, H.; Pytowski, L.; Grovenor, C.; Vaux, D.J. Formation of a nucleoplasmic reticulum requires de novo assembly of nascent phospholipids and shows preferential incorporation of nascent lamins. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Broers, J.L.V.; Peeters, E.A.G.; Kuijpers, H.J.H.; Endert, J.; Bouten, C.V.C.; Oomens, C.W.J.; Baaijens, F.P.T.; Ramaekers, F.C.S. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum. Mol. Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef] [Green Version]
- Houben, F.; Willems, C.H.M.P.; Declercq, I.L.J.; Hochstenbach, K.; Kamps, M.A.; Snoeckx, L.H.E.H.; Ramaekers, F.C.S.; Broers, J.L.V. Disturbed nuclear orientation and cellular migration in A-type lamin deficient cells. Biochim. Biophys. Acta. Mol. Cell Res. 2009, 1793, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamiello, C.; Kamps, M.A.F.; Van den Wijngaard, A.; Verstraeten, V.L.R.M.; Baaijens, F.P.T.; Broers, J.L.V.; Bouten, C.C.V. Soft substrates normalize nuclear morphology and prevent nuclear rupture in fibroblasts from a laminopathy patient with compound heterozygous LMNA mutations. Nucleus 2013, 4, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatch, E.M.; Hetzer, M.W. Nuclear envelope rupture is induced by actin-based nucleus confinement. J. Cell Biol. 2016, 215, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior—life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; Perez, D.; White, E. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 1996, 135, 1441–1455. [Google Scholar] [CrossRef]
- Galiová, G.; Bártová, E.; Raška, I.; Krejčí, J.; Kozubek, S. Chromatin changes induced by lamin A/C deficiency and the histone deacetylase inhibitor trichostatin A. Eur. J. Cell Biol. 2008, 87, 291–303. [Google Scholar] [CrossRef]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C–deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef]
- Ognibene, A.; Sabatelli, P.; Petrini, S.; Squarzoni, S.; Riccio, M.; Santi, S.; Villanova, M.; Palmeri, S.; Merlini, L.; Maraldi, N. Nuclear changes in a case of X-linked Emery-Dreifuss muscular dystrophy. Muscle Nerve. 1999, 22, 864–869. [Google Scholar] [CrossRef]
- Scaffidi, P. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- De Vos, W.H.; Houben, F.; Hoebe, R.A.; Hennekam, R.; van Engelen, B.; Manders, E.M.M.; Ramaekers, F.C.S.; Broers, J.L.V.; Van Oostveldt, P. Increased plasticity of the nuclear envelope and hypermobility of telomeres due to the loss of A–type lamins. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Lammerding, J. Lamins at a glance. J. Cell Sci. 2012, 125, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Davidson, P.M.; Lammerding, J. Broken nuclei—lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, I.; Kind, J. Spatial chromatin organization and gene regulation at the nuclear lamina. Curr. Opin. Genet. Dev. 2019, 55, 19–25. [Google Scholar] [CrossRef]
- Newport, J.W.; Wilson, K.L.; Dunphy, W.G. A lamin-independent pathway for nuclear envelope assembly. J. Cell Biol. 1990, 111, 2247–2259. [Google Scholar] [CrossRef] [Green Version]
- Champion, L.; Linder, M.I.; Kutay, U. Cellular reorganization during mitotic entry. Trends Cell Biol. 2017, 27, 26–41. [Google Scholar] [CrossRef]
- Broers, J.L.; Machiels, B.M.; van Eys, G.J.; Kuijpers, H.J.; Manders, E.M.; van Driel, R.; Ramaekers, F.C. Dynamics of the nuclear lamina as monitored by GFP-tagged A-type lamins. J. Cell Sci. 1999, 112, 3463–3475. [Google Scholar]
- Ingber, D.E.; Tensegrity, I. Cell structure and hierarchical systems biology. J. Cell Sci. 2003, 116, 1157–1173. [Google Scholar] [CrossRef] [Green Version]
- Dahl, K.N.; Kahn, S.M.; Wilson, K.L.; Discher, D.E. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J. Cell Sci. 2004, 117, 4779–4786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wintner, O.; Hirsch-Attas, N.; Schlossberg, M.; Brofman, F.; Friedman, R.; Kupervaser, M.; Kitsberg, D.; Buxboim, A. A unified linear viscoelastic model of the cell nucleus defines the mechanical contributions of lamins and chromatin. Adv. Sci. 2020, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef]
- Corne, T.D.J.; Sieprath, T.; Vandenbussche, J.; Mohammed, D.; te Lindert, M.; Gevaert, K.; Gabriele, S.; Wolf, K.; De Vos, W.H. Deregulation of focal adhesion formation and cytoskeletal tension due to loss of A-type lamins. Cell Adh. Migr. 2017, 11, 447–463. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.H.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [Green Version]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- Isermann, P.; Lammerding, J. Consequences of a tight squeeze: Nuclear envelope rupture and repair. Nucleus 2017, 8, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Houthaeve, G.; Robijns, J.; Braeckmans, K.; De Vos, W.H. Bypassing Border Control: Nuclear Envelope Rupture in Disease. Physiology 2018, 33, 39–49. [Google Scholar] [CrossRef]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; Te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Bhattacharyya, M.; Sengupta, K. Implications and assessment of the elastic behavior of lamins in laminopathies. Cells 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, R.; Hegele, R.A. Complex effects of laminopathy mutations on nuclear structure and function. Clin. Genet. 2019, 95, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Luxton, G.W.; Gomes, E.R.; Folker, E.S.; Worman, H.J.; Gundersen, G.G. TAN lines: A novel nuclear envelope structure involved in nuclear positioning. Nucleus 2011, 2, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.L.; Foisner, R. Lamin-binding proteins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego-Pinto, J.; Jegou, T.; Osorio, D.S.; Auradé, F.; Gorjánácz, M.; Koch, B.; Mattaj, I.W.; Gomes, E.R. Samp1 is a component of TAN lines and is required for nuclear movement. J. Cell Sci. 2012, 125, 1099–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoku, S.; Zhu, R.; Kutscheidt, S.; Fackler, O.T.; Gundersen, G.G. Reinforcing the LINC complex connection to actin filaments: The role of FHOD1 in TAN line formation and nuclear movement. Cell Cycle 2015, 14, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.A.; Harris, N.J.; Willey, P.T.; Woolums, B.M.; Wang, Y.; McQuown, A.J.; Schoenhofen, A.; Worman, H.J.; Dauer, W.T.; Gundersen, G.G.; et al. TorsinA controls TAN line assembly and the retrograde flow of dorsal perinuclear actin cables during rearward nuclear movement. J. Cell Biol. 2017, 216, 657–674. [Google Scholar] [CrossRef]
- Van Loosdregt, I.A.E.W.; Kamps, M.A.F.; Oomens, C.W.J.; Loerakker, S.; Broers, J.L.V.; Bouten, C.V.C. Lmna knockout mouse embryonic fibroblasts are less contractile than their wild-type counterparts. Integr. Biol. 2017, 9, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.J.; Fischer, M.; Jabre, S.; Moog, S.; Mamchaoui, K.; Butler-Browne, G.; Coirault, C. Lamin mutations cause increased YAP nuclear entry in muscle stem cells. Cells 2020, 9, 816. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.S.; Lammerding, J. Causes and consequences of nuclear envelope alterations in tumour progression. Eur. J. Cell Biol. 2016, 95, 449–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K.; Lammerding, J. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 2011, 23, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.-W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihalainen, T.O.; Aires, L.; Herzog, F.A.; Schwartlander, R.; Moeller, J.; Vogel, V. Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat. Mater. 2015, 14, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Pieuchot, L.; Marteau, J.; Guignandon, A.; Dos Santos, T.; Brigaud, I.; Chauvy, P.-F.; Cloatre, T.; Ponche, A.; Petithory, T.; Rougerie, P.; et al. Curvotaxis directs cell migration through cell-scale curvature landscapes. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Pfeifer, C.R.; Cho, S.; Discher, D.E.; Irianto, J. Nuclear mechanosensing. Emerg. Top. Life Sci. 2018, 2, 713–725. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Breaching the barrier—the nuclear envelope in virus infection. J. Mol. Biol. 2016, 428, 1949–1961. [Google Scholar] [CrossRef]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Mezaki, N.; Miura, T.; Ogaki, K.; Eriguchi, M.; Mizuno, Y.; Komatsu, K.; Yamazaki, H.; Suetsugi, N.; Kawajiri, S.; Yamasaki, R.; et al. Duplication and deletion upstream of LMNB1 in autosomal dominant adult-onset leukodystrophy. Neurol. Genet. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, E.; Sirchia, F.; Bosco, M.; Sobreira, N.L.M.; Grosso, E.; Brussino, A.; Brusco, A. A novel case of Greenberg dysplasia and genotype–phenotype correlation analysis for LBR pathogenic variants: An instructive example of one gene-multiple phenotypes. Am. J. Med. Genet. Part A 2019, 179, 306–311. [Google Scholar] [CrossRef]
- Hegele, R.A.; Cao, H.; Liu, D.M.; Costain, G.A.; Charlton-Menys, V.; Wilson Rodger, N.; Durrington, P.N. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am. J. Hum. Genet. 2006, 79, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tienen, F.H.J.; Lindsey, P.J.; Kamps, M.A.F.; Krapels, I.P.; Ramaekers, F.C.S.; Brunner, H.G.; van den Wijngaard, A.; Broers, J.L.V. Assessment of fibroblast nuclear morphology aids interpretation of LMNA variants. Eur. J. Hum. Genet. 2018, 27, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaudain, A.; Vantyghem, M.-C.; Guerci, B.; Heécart, A.-C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.-H.; Donadille, B.; Lebbeé, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Vashisth, M.; Abbas, A.; Greenberg, R.A.; Prosser, B.L.; Discher Correspondence, D.E.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; et al. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell 2019, 49, 920–935. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Pfeifer, C.R.; Zhu, K.; Irianto, J.; Liu, D.; Pannell, K.; Chen, E.J.; Dooling, L.J.; Tobin, M.P.; Wang, M.; et al. Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycle. J. Cell Biol. 2019, 218, 2545–2563. [Google Scholar] [CrossRef] [PubMed]
- De Vos, W.H.; Houben, F.; Kamps, M.; Malhas, A.; Verheyen, F.; Cox, J.; Manders, E.M.M.; Verstraeten, V.L.R.M.; van Steensel, M.A.M.; Marcelis, C.L.M.; et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol. Genet. 2011, 20, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Booth-Gauthier, E.A.; Du, V.; Ghibaudo, M.; Rape, A.D.; Dahl, K.N.; Ladoux, B. Hutchinson-Gilford progeria syndrome alters nuclear shape and reduces cell motility in three dimensional model substrates. Integr. Biol. 2013, 5, 569–577. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 697, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Tamiello, C.; Halder, M.; Kamps, M.A.; Baaijens, F.P.; Broers, J.L.; Bouten, C.V. Cellular strain avoidance is mediated by a functional actin cap—observations in an Lmna-deficient cell model. J. Cell Sci. 2017, 130, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Louhghalam, A.; Lee, G.; Schafer, B.W.; Wirtz, D.; Kim, D.H. Nuclear lamin A/C harnesses the perinuclear apical actin cables to protect nuclear morphology. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Schwartz, C.; Fischer, M.; Mamchaoui, K.; Bigot, A.; Lok, T.; Verdier, C.; Duperray, A.; Michel, R.; Holt, I.; Voit, T.; et al. Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci. Rep. 2017, 7, 1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.M.; Rodriguez, E.C.; King, M.C. Ablation of SUN2-containing LINC complexes drives cardiac hypertrophy without interstitial fibrosis. Mol. Biol. Cell 2019, 30, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Brull, A.; Azibani, F.; Benarroch, L.; Chikhaoui, K.; Stewart, C.L.; Medalia, O.; Ben Yaou, R.; Bonne, G. Lamin A/C assembly defects in LMNA-congenital muscular dystrophy is responsible for the increased severity of the disease compared with Emery–Dreifuss muscular dystrophy. Cells 2020, 9, 844. [Google Scholar] [CrossRef] [Green Version]
- Dechat, T.; Shimi, T.; Adam, S.A.; Rusinol, A.E.; Andres, D.A.; Spielmann, H.P.; Sinensky, M.S.; Goldman, R.D. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc. Natl. Acad. Sci. USA 2007, 104, 4955–4960. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, O.A.; Alvarez-Reyes, M.; Bridger, J.M.; Broers, J.L.V.; Ramaekers, F.C.S.; Wehnert, M.; Morris, G.E.; Whitfield, W.G.F.; Hutchison, C.J. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 2001, 114, 2577–2590. [Google Scholar]
- Koch, A.J.; Holaska, J.M. Emerin in health and disease. Semin. Cell Dev. Biol. 2014, 29, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Östlund, C.; Chang, W.; Gundersen, G.G.; Worman, H.J. Pathogenic mutations in genes encoding nuclear envelope proteins and defective nucleocytoplasmic connections. Exp. Biol. Med. 2019, 244, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Foisner, R. The structural and gene expression hypotheses in laminopathic diseases—not so different after all. Mol. Biol. Cell 2019, 30, 1786–1790. [Google Scholar] [CrossRef]
- Broers, J.L.V.; Ramaekers, F.C.S. The role of the nuclear lamina in cancer and apoptosis. Adv. Exp. Med. Biol. 2014, 773, 27–48. [Google Scholar] [CrossRef]
- Hudson, M.E.; Pozdnyakova, I.; Haines, K.; Mor, G.; Snyder, M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc. Natl. Acad. Sci. USA 2007, 104, 17494–17499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, N.D.; Cox, T.R.; Rahman-Casañs, S.F.; Smits, K.; Przyborski, S.A.; van den Brandt, P.; van Engeland, M.; Weijenberg, M.; Wilson, R.G.; de Bruïne, A.; et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS ONE 2008, 3, e2988. [Google Scholar] [CrossRef] [Green Version]
- Tilli, C.M.L.J.; Ramaekers, F.C.S.; Broers, J.L.V.; Hutchison, C.J.; Neumann, H.A.M. Lamin expression in normal human skin, actinic keratosis, squamous cell carcinoma and basal cell carcinoma. Br. J. Dermatol. 2003, 148, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Coradeghini, R.; Barboro, P.; Rubagotti, A.; Boccardo, F.; Parodi, S.; Carmignani, G.; D’Arrigo, C.; Patrone, E.; Balbi, C. Differential expression of nuclear lamins in normal and cancerous prostate tissues. Oncol. Rep. 2006, 15, 609–613. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, R.; Cho, K.R.; Thomas, D.G.; Gossner, G.; Liu, J.R.; Giordano, T.J.; Shedden, K.A.; Misek, D.E.; Lubman, D.M. Differential protein mapping of ovarian serous adenocarcinomas: Identification of potential markers for distinct tumor stage. J. Proteome Res. 2009, 8, 1452–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belt, E.J.T.; Fijneman, R.J.A.; Van Den Berg, E.G.; Bril, H.; Delis-Van Diemen, P.M.; Tijssen, M.; Van Essen, H.F.; De Lange-De Klerk, E.S.M.; Beliën, J.A.M.; Stockmann, H.B.A.C.; et al. Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence. Eur. J. Cancer 2011, 47, 1837–1845. [Google Scholar] [CrossRef] [Green Version]
- Venables, R.S.; McLean, S.; Luny, D.; Moteleb, E.; Morley, S.; Quinlan, R.A.; Lane, E.B.; Hutchison, C.J. Expression of individual lamins in basal cell carcinomas of the skin. Br. J. Cancer 2001, 84, 512–519. [Google Scholar] [CrossRef]
- Agrelo, R.; Setien, F.; Espada, J.; Artiga, M.J.; Rodriguez, M.; Pérez-Rosado, A.; Sanchez-Aguilera, A.; Fraga, M.F.; Piris, M.A.; Esteller, M. Inactivation of the Lamin A/C gene by CpG island promoter hypermethylation in hematologic malignancies, and its association with poor survival in nodal diffuse large B-cell lymphoma. J. Clin. Oncol. 2005, 23, 3940–3947. [Google Scholar] [CrossRef]
- Moss, S.F.; Krivosheyev, V.; De Souza, A.; Chin, K.; Gaetz, H.P.; Chaudhary, N.; Worman, H.J.; Holt, P.R. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 1999, 45, 723–729. [Google Scholar] [CrossRef]
- Broers, J.L.V.; Raymond, Y.; Rot, M.K.; Kuijpers, H.; Wagenaar, S.S.; Ramaekers, F.C.S. Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am. J. Pathol. 1993, 143, 211–220. [Google Scholar] [CrossRef]
- Capo-Chichi, C.D.; Cai, K.Q.; Smedberg, J.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.X. Loss of A-type lamin expression compromises nuclear envelope integrity in breast cancer. Chin. J. Cancer 2011, 30, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wu, L.; Weng, D.; Xu, D.; Geng, J.; Zhao, F. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De las Heras, J.I.; Batrakou, D.G.; Schirmer, E.C. Cancer biology and the nuclear envelope: A convoluted relationship. Semin. Cancer Biol. 2013, 23, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, B.M.; Broers, J.L.; Raymond, Y.; de Ley, L.; Kuijpers, H.J.; Caberg, N.E.; Ramaekers, F.C. Abnormal A-type lamin organization in a human lung carcinoma cell line. Eur. J. Cell Biol. 1995, 67, 328–335. [Google Scholar]
- Sun, S.; Xu, M.Z.; Poon, R.T.; Day, P.J.; Luk, J.M. Circulating lamin B1 (LMNB1) biomarker detects early stages of liver cancer in patients. J. Proteome Res. 2010, 9, 70–78. [Google Scholar] [CrossRef]
- Fu, Y.; Chin, L.K.; Bourouina, T.; Liu, A.Q.; Vandongen, A.M.J. Nuclear deformation during breast cancer cell transmigration. Lab. Chip 2012, 12, 3774–3778. [Google Scholar] [CrossRef]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.D.P.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Kornfeind, E.M.; Visalli, R.J. Human herpesvirus portal proteins: Structure, function, and antiviral prospects. Rev. Med. Virol. 2018, 28, 1–15. [Google Scholar] [CrossRef]
- Kobiler, O.; Brodersen, P.; Taylor, M.P.; Ludmir, E.B.; Enquist, L.W. Herpesvirus replication compartments originate with single incoming viral genomes. MBio 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Cibulka, J.; Fraiberk, M.; Forstova, J. Nuclear actin and lamins in viral infections. Viruses 2012, 4, 325–347. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.E.; Liang, L.; Baines, J.D. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes UL31 and UL34. J. Virol. 2004, 78, 5564–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.C.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The prolyl ssomerase Pin1 promotes the herpesvirus-induced phosphorylation-dependent disassembly of the nuclear lamina required for nucleocytoplasmic egress. PLoS Pathog. 2016, 12, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, A.; Poyzer, C.; Roller, R. Extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects lamina disruption and nuclear egress. J. Virol. 2016, 90, 10738–10751. [Google Scholar] [CrossRef] [Green Version]
- Mou, F.; Wills, E.G.; Park, R.; Baines, J.D. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus UL34-encoded protein to the inner nuclear membrane. J. Virol. 2008, 82, 8094–8104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roller, R.J.; Bjerke, S.L.; Haugo, A.C.; Hanson, S. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J. Virol. 2010, 84, 3921–3934. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, Y.; Wu, S.; Pan, S.; Zhou, C.; Ma, Y.; Ru, Y.; Dong, S.; He, B.; Zhang, C.; et al. P32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J. Biol. Chem. 2014, 289, 35795–35805. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes simplex virus 1 induces phosphorylation and reorganization of lamin A/C through the γ1 34.5 protein that facilitates nuclear egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Chattaraj, S. Entry, infection, replication, and egress of human polyomaviruses: An update. Can. J. Microbiol. 2017, 63, 193–211. [Google Scholar] [CrossRef]
- Butin-Israeli, V.; Ben-nun-Shaul, O.; Kopatz, I.; Adam, S.A.; Shimi, T.; Goldman, R.D.; Oppenheim, A. Simian virus 40 induces lamin A/C fluctuations and nuclear envelope deformation during cell entry. Nucleus 2011, 2, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Suzuki, T.; Sunden, Y.; Orba, Y.; Kose, S.; Imamoto, N.; Takahashi, H.; Tanaka, S.; Hall, W.W.; Nagashima, K.; et al. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: Role in nuclear egress of viral particles. EMBO Rep. 2005, 6, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Panou, M.M.; Prescott, E.L.; Hurdiss, D.L.; Swinscoe, G.; Hollinshead, M.; Caller, L.G.; Morgan, E.L.; Carlisle, L.; Müller, M.; Antoni, M.; et al. Agnoprotein is an essential egress factor during BK Polyomavirus infection. Int. J. Mol. Sci. 2018, 19, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xu, K.; Wei, D.; Wu, W.; Yang, K.; Yuan, M. Baculovirus infection induces disruption of the nuclear lamina. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Marr, A.K.; Garcin, P.; Pante, N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 2011, 85, 4863–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porwal, M.; Cohen, S.; Snoussi, K.; Popa-Wagner, R.; Anderson, F.; Dugot-Senant, N.; Wodrich, H.; Dinsart, C.; Kleinschmidt, J.A.; Panté, N.; et al. Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog. 2013, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Panté, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef]
- Mäntylä, E.; Niskanen, E.A.; Ihalainen, T.O.; Vihinen-Ranta, M. Reorganization of nuclear pore complexes and the lamina in late-stage parvovirus infection. J. Virol. 2015, 89, 11706–11710. [Google Scholar] [CrossRef] [Green Version]
- De Noronha, C.M.C.; Sherman, M.P.; Lin, H.W.; Cavrois, M.V.; Moir, R.D.; Goldman, R.D.; Greene, W.C. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 2001, 294, 1105–1108. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiekema, M.; van Zandvoort, M.A.M.J.; Ramaekers, F.C.S.; Broers, J.L.V. Structural and Mechanical Aberrations of the Nuclear Lamina in Disease. Cells 2020, 9, 1884. https://doi.org/10.3390/cells9081884
Stiekema M, van Zandvoort MAMJ, Ramaekers FCS, Broers JLV. Structural and Mechanical Aberrations of the Nuclear Lamina in Disease. Cells. 2020; 9(8):1884. https://doi.org/10.3390/cells9081884
Chicago/Turabian StyleStiekema, Merel, Marc A. M. J. van Zandvoort, Frans C. S. Ramaekers, and Jos L. V. Broers. 2020. "Structural and Mechanical Aberrations of the Nuclear Lamina in Disease" Cells 9, no. 8: 1884. https://doi.org/10.3390/cells9081884
APA StyleStiekema, M., van Zandvoort, M. A. M. J., Ramaekers, F. C. S., & Broers, J. L. V. (2020). Structural and Mechanical Aberrations of the Nuclear Lamina in Disease. Cells, 9(8), 1884. https://doi.org/10.3390/cells9081884