1. Introduction
Immune therapy represents a novel type of treatment, which boosts the body’s immune system to recognize and eliminate pathogens or abnormal cells, as in cancers, and thus establish a defense memory. Therefore, immune therapy has become a central pillar in cancer treatment, either as first-line therapy or as an additive to classical chemotherapy. In the late 19th century, cancer immune therapy was initiated by Coley’s toxin, a mixture of killed streptococcus bacteria, then progressed by the use of systemic cytokines, such as interleukin (IL)-2, and evolved to the more recently approved checkpoint inhibitors anti-CTLA-4 and anti-PD-1/PD-L1 [
1].
Tumor cells acquire three major features to evade immune recognition in order to grow and progress: the ability to 1. thrive in a chronically inflamed, immunosuppressive microenvironment; 2. evade immune recognition by the host; 3. actively suppress the host’s immune response [
1].
The (advanced) tumor microenvironment consists of subsets of immunosuppressive cells, and macrophages often play a central in cancer immune suppression. Usually, macrophages are the first line of defense against of the innate immune system to fight pathogens and to clearance foreign, aged, and damaged cells [
2]. In chronic diseases, especially cancer and fibrosis, macrophages are usually polarized towards an immunosuppressive, alternative (M2-type) macrophage phenotype. It is mainly the cancer cells and the tumor associated stroma that promote macrophage repolarization towards the M2-type, i.e., tumor-associated macrophages (TAM). TAM dampen Th1 T cell or cytotoxic immune reactions, promote the rise of regulatory T-cells (Treg), or of other immune suppressive cells of the innate immune system, such as myeloid derived suppressor cells [
2]. TAM are the source of various pro-angiogenic, pro-fibrotic, and T cell suppressive cytokines, e.g., vascular endothelial growth factor (VEGF) or transforming growth factor beta 1 (TGFβ1) [
2].
Besides cancer, M2-type macrophages also play an important role in the development of organ fibrosis, which is considered the result of “wounds that do not heal” [
3,
4,
5,
6]. In the liver, fibrosis, and its end-stage cirrhosis, results from continuous inflammation, e.g., due to viral hepatitis, alcoholic or nonalcoholic steatohepatitis, or autoimmune liver diseases. Here, M2-type macrophages can subdue the chronic M1-type inflammation, while at the same time they promote scarring [
7,
8]. Cirrhosis gives rise to life-threatening complications linked to portal hypertension and liver failure. Importantly, cirrhosis is tightly linked to the development of liver cancer (hepatocellular carcinoma, HCC). Thus, the vast majority of HCC evolve in cirrhotic livers [
9,
10]. This association can be explained by the abnormal stroma of cirrhosis, favoring M2-type macrophages and the resulting immune suppression.
In liver fibrosis, M2 polarized macrophages mirror, therefore, many characteristics and qualities of TAM. Although the role of macrophages and their phenotype in liver fibrosis is complex, M2-type macrophages are pro-fibrotic in fibrosis progression and tumor stromal remodeling, and in cirrhotic livers, TAM by far outnumber other macrophage phenotypes and are correlated with poor prognosis in HCC [
11,
12,
13].
Notably, even with a certain phenotype macrophages maintain their plasticity. Thus, M2-type macrophages can be re-educated to resume their original anti-cancer and anti-fibrotic M1-type polarization by small interfering RNA (siRNA) directed to macrophage receptors [
14,
15,
16]. Such siRNA or DNA antisense strategies have also been effective in repolarizing the profibrotic M2 phenotype in mouse models of liver fibrosis [
3,
17,
18,
19].
Although RNA interference can be a powerful tool to target relevant transcripts in vivo, naked siRNA possesses unfavorable pharmacokinetics: After parenteral administration, siRNA is prone to rapid kidney clearance caused by its low molecular weight as well as immanent degradation by nucleases of the blood stream. Due to its highly negative charge, naked siRNA cannot penetrate cell membranes but triggers unspecific (innate) immune reactions [
20]. Therefore, a suitable carrier system is required to overcome these limitations. Such system should also secure cell-specific delivery of siRNA, here to M2-type macrophages, to reduce the risk of off-target effects while achieving therapeutically relevant siRNA concentrations in the target cells.
To address these needs, we have established a block copolymer-based delivery system derived from reactive precursor polymers that self-assemble into micelles whose cores are then crosslinked and switched from hydrophobic to polycationic, allowing for efficient siRNA encapsulation [
20,
21,
22]. Compared to other established siRNA delivery vehicles, these nanogels provide favorable characteristics for in vivo delivery, as demonstrated by us before [
20,
23,
24,
25,
26]. Since M2 polarized macrophages overexpress the mannose receptor CD206, we have recently extended our non-targeted cationic nanohydrogel particle platform (NonNP) by applying heterotelechelic block copolymers with α-mannosyl end groups, resulting in α-mannose nanohydrogels (ManNP) [
23]. In vitro, NonNP and ManNP showed no cytotoxicity and were preferentially taken up by the M2 polarized macrophages and demonstrated functional siRNA delivery by M2-specific knockdown of CSF-1 receptor (CSF-1R) mRNA, a relevant target in M2-type macrophages [
14,
23].
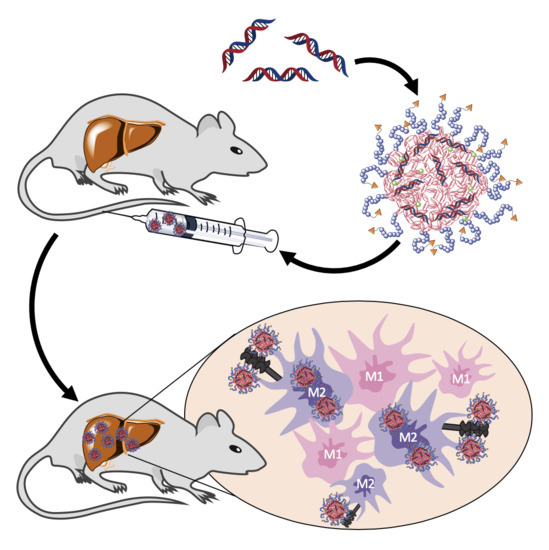
In this study, we applied ManNP and NonNP in vivo to mice with active liver fibrosis to assess their M2 macrophage-specific delivery. Dual near infrared dye labeling allowed in vivo monitoring of siRNA and NP delivery into organs and ex vivo immunohistochemical colocalization studies in the liver. We show that ManNP specifically targeted hepatic M2-type macrophages and demonstrated no organ or cellular toxicity, qualifying them as promising carriers for siRNA-directed macrophage repolarization in liver cancer and fibrosis.
2. Materials and Methods
2.1. Fabricationof α-Mannosyl Modified Cationic Nanohydrogel Particles
The fabrication of non-functionalized cationic nanohydrogel particles was performed as previously reported in detail [
23]. In brief, the synthesis of ManNP was conducted equivalent to NonNPs and is described below for fluorescent labeled particles with Oregon Green 488. For in vivo biodistribution studies, (Non-)ManNPs were fabricated in analogy but using the near infrared dye NIR 800RS cadaverine, as fluorescent dye instead of Oregon Green 488.
All steps were conducted under argon atmosphere. In dry DMSO Man-P(MEO
3MA)
18-
b-P(PFPMA)
30 (40 mg; 3.24 µmol or 97.2 µmol reactive ester) as α-mannosyl end group modified amphiphilic block copolymer was dissolved (4 mL; 10 g/L) and treated with triethylamine (7.3 µL; 10% of total 0.54 mmol) and the corresponding fluorescent dye (89.8 µL of a 2.5 g/L stock solution in anhydrous DMSO; 0.45 µmol). The reaction mixture was vigorously stirred at room temperature for 16 h. Then, spermine (9.2 mg; 45.2 µmol) and once more triethylamine (68 µL; 90% of total 0.54 mmol) were added to initiate crosslinking. After stirring for additional 24 h at 50 °C, a sample was taken for
19F-NMR to determine complete conversion of the reactive ester units. To ensure further full conversion of all PFPMA units, spermine (18.3 mg; 90.5 µmol) was added once more, followed by 18 h stirring. Afterwards, the reaction mixture was dialyzed against Millipore water for several days, changing the water twice per day. The mixture was lyophilized and cationic nanohydrogel particles could be obtained as orange powder (25.9 mg; 2.63 µmol; 81%). For the detailed description of the synthesis and physiochemical analysis of the cationic nanohydrogel particles, refer to our previous work [
23].
2.2. Loading of (Non-)ManNP with siRNA
SiRNA was complexed inside the NonNP at a weight ratio of 10:1 for 20 min at room temperature. SiRNAs were purified by high-performance liquid chromatography. A Cy5 labeled scrambled siRNA was used with the dye attached to the 5′ by a C6 amino spacer. It was purchased from bearing Dharmacon (Freiburg, Germany) with the following sequence:
5′-Cy5 GCAUCUGGCUUAAGGUGAAUU-3′
5′-PGUAUCUCUUCAUAGCCUUAUU-3′
2.3. Cell Culture: Cell Lines
The cell culture work has been performed as described by us in detail previously [
23]. In brief, THP-1 monocytes, RAW macrophages or SVEC4-10 endothelial cells, and HepG2 human hepatocytes were cultured in RPMI Media 1640 (Sigma-Aldrich, Taufkirchen, Germany) or Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich, Taufkirchen, Germany) or Eagle’s Minimum Essential Medium (EMEM) (Sigma-Aldrich, Taufkirchen, Germany) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1% l-glutamine, and 50 pM β-mercaptoethanol (Gibco; 31350–010) for THP-1 in 5% CO2 at 37 °C, respectively. The cells were passaged just before reaching confluency. For cell detachment, THP-1 and RAW cells were incubated in PBS buffer, without calcium and magnesium, supplemented with 10 mm EDTA, for 15 min on ice and then carefully detached by cell scraping, while Svec4-10 and HepG2 could be trypsinized with Trypsin-EDTA solution (Sigma-Aldrich, Taufkirchen, Germany). For differentiation into macrophages, THP-1 monocytes were incubated with 150 nM phorbol 12-myristate 13-acetate (Sigma-Aldrich, Taufkirchen, Germany) for 24 h followed by 12 h incubation in RPMI medium.
2.4. Cell Culture: Primary Macrophages—Bone Marrow Derived Macrophages (BMDM)
As described before in detail [
23], bone marrow cells were extracted from the bone marrow of mice and matured in the presence of a macrophage colony-stimulating factor (M-CSF), a lineage-specific growth factor, into primary macrophages. Bone marrow was extracted from 6 to 9 weeks old Balb/c mice. In brief, mice were sacrificed by cervical dislocation, and femurs and tibias were isolated under sterile conditions. A 21G needle and a 10 mL syringe served to rinse out the marrow into cold PBS substituted with 2% heat inactivated FBS and 1% penicillin/streptomycin. Afterwards, cells were passed through a 70 µm cell strainer for cell separation and purification. Remaining red blood cells were lysed with red cell lysis buffer (eBioscience, San Diego, CA, USA). The resulting bone marrow cells were resuspended in Iscove’s Modified Dulbecco’s Medium (IMDM), 10% FBS, 1% penicillin/streptomycin, 1% L-glutamine containing 25 ng/mL monocyte colony-stimulating factor (M-CSF, ImmunoTools, Friesoythe, Germany). Approximately 5 million cells were plated out into bacterial culture plate in 10 mL (Sigmal-Aldrich/Merck, Darmstadt, Germany). On the following days, half of the media was replaced and substituted with fresh IMDM containing the same supplements. Cells were incubated for an additional 4 days in the presence of M-CSF for further differentiation into mature primary macrophages. M2- or M1-phenotype was obtained, when primary macrophages were incubated with fully supplemented IMDM containing 20 ng/mL IL-4 and IL-13 or 50 ng/mL interferon-γ and 0.1 µg mL
−1 LPS (all from ImmunoTools, Friesoythe, Germany), respectively, for 24 h. Sub-culturing was performed as described above for THP-1 and RAW cells.
2.5. In Vitro Cytotoxicity
In vitro cytotoxicity of scrambled siRNA-loaded (non-)ManNP on THP-1 and SVEC4-10 was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium-bromide) assay as described before [
23]. In brief, cells were plated out into 96-well plates at a density of 6000–7000 cells per well and incubated in a fully supplemented medium. After 24 h, increasing concentrations of siRNA/NP (10:1 weight-to-weight ratio of (non-)ManNP to siRNA) corresponding to 5, 10, 20, 40, 60, 80, 100, 200, and 300 nm siRNA were added. After 48 h of incubation, 20 µL of MTT in PBS (4 mg/mL) was added to each well and incubated for 5 h. The medium was then carefully removed and 150 µL of DMSO was added. After full solubilization, optical density (OD) was measured on an Infinite M200Pro spectrofluorometer at 570 nm (TECAN, Männerdorf, Switzerland).
2.6. In Vitro Cellular Uptake of the siRNA-Loaded Carriers by Fluorescent Activated Cell Sorting Flow Cytometry (FACS)
Cells were plated out in 12-well culture plates at a density of 130,000 cells per well and incubated for 24 h. Cells were incubated with a final concentration of 25, 50, and 100 nM Cy5-labeled scrambled siRNA complexed with (Non-)ManNP (10:1 weight-to-weight ratio of (Non-)ManNP: siRNA) for 1 h at 37 °C. For normalization of potential autofluorescence of siRNA and nanoparticles, cells were incubated with a final concentration of 100 nM unlabeled scrambled siRNA complexed with unlabeled (Non-)ManNP (10:1 weight-to-weight ratio of (Non-)ManNP: siRNA) for 1 h in the cell incubator before FACS analysis. Control cells were incubated with equal volumes of PBS for 1 h at 37 °C. After 1 h incubation, culture medium was poured off, and the cells were repetitively washed with PBS. Detachment of the cells was performed as described above. Cells were then also stained with a viability dye (Fixable Viability Dye eFluor® 506, eBioscience, San Diego, CA, USA) according to the manufacture’s protocol and stored at 4 °C under the exclusion of light. Finally, cells could be analyzed by flow cytometry (BD FACS Canto II, BD Bioscience, Canada, Mississauga) on the same day. 50,000 cells were measured per sample. The obtained data were analyzed with Flowing Software 2.5.0 (Perttu Terho, Turku Centre for Biotechnology, Finland).
2.7. Fibrosis Model
The conducted animal studies followed the approval of the local ethics committee on animal care (number 23 177-07/G 17-1-030, Government of Rhineland Palatinate, Germany). 8–10-week-old female Balb/c mice (body weight ~20–25 g) were bought from Charles River (Sulzfeld, Germany) and kept according the guidelines of the institute and the local government (12 h light-dark cycles at 25 °C and 40–60% humidity with humane care, access to regular chow and water ad libitum). Carbon tetrachloride (CCl
4) (Sigma-Aldrich, St. Louis, US) diluted 1:1 in mineral oil (Sigma-Aldrich, St. Louis, US) was administered by oral gavage 3 times per week following an escalating dose protocol (first dose 0.875 mL/kg; 1.75 mL/kg week 1; 2.5 mL/kg week 2, 3, and 4), as reported in detail before [
26]. Mice gavaged with pure mineral oil not containing CCl4 served as non-fibrotic healthy controls. At the described time points, mice were sacrificed by cervical dislocation.
2.8. In Vivo Imaging of Near Infrared (NIR) Labeled siRNA and Nanogel Particles
At week 4 of CCl4 treatment, mice (n = 5 per group) were anesthetized with isoflurane gas and treated via tail vain injection with 2 mg/kg Cy5-labeled scrambled siRNA loaded into NIR dye RS800 labelled (Non-)ManNP (10:1 weight-to-weight ratio of (Non-)ManNP: siRNA) for in vivo NIR fluorescence imaging by an IVIS (in vivo imaging spectrum) system (Caliper Life Sciences, Hopkinton, US). At the given time points, mice were anesthetized with isoflurane gas and transferred into the imaging chamber according the manufactures protocol. Picture integration time of the fluorescence source was set to 4 s. Recording parameters were set for excitation at 745 nm and emission at 800 nm to visualize IRDye RS800 (near infrared dye) labeled nanohydrogel particles, while for detecing Cy5-dye-labeled scsiRNA they were adjusted to excitation at 640 nm and emission at 700 nm.
2.9. Ex Vivo Imaging of Organs
24 h after Cy5-scsiRNA-loaded (Non-)ManNP injection, mice were sacrificed, and organs were harvested and put into the imaging chamber. Image setup was adjusted in analogy to the settings described above. Ex vivo signal quantification of Cy5-scsiRNA and NIR-NonNP in the isolated organs could then be quantified by using Live Image software from PerkinElmer. Organs were carefully circumscribed by region-of-interests (ROIs), and then their fluorescence was quantified as the average of area-normalized radiant efficiency.
2.10. Production of Liver Single Cell Suspensions
After imaging, livers were processed according to obtain single cell suspensions following the manufactures’ protocol (Milteny, Bergisch Gladbach, Germany) as described by us previously [
25,
26]. Briefly, livers were washed with PBS and, after extraction of the gall bladder, digested with 5000 U/mL of collagenase IV (C5138, Sigma-Aldrich) in Krebs-Ringer-Buffer, pH 7.4. The livers were carefully minced with a gentleMACS™ dissociator (Milteny, Germany, Bergisch Gladbach) and incubated for 30 min at 37 °C. This step was repeated once more before the cell suspension could be filtered over a 100 µm cell strainer to remove undigested tissue clumps. The filtered cell suspension was afterwards centrifuged at 300×
g for 10 min at 4 °C, and thus, parenchymal and non-parenchymal cells could be collected. The supernatant was poured off, and the remaining cell pellet was then resuspended in 5 mL 1× Red Blood Cell Lysis Solution (Milteny, Germany, Bergisch Gladbach) for 5 min at RT to remove the remaining erythrocytes. Finally, remaining live cells were washed 3 times with PBS and further processed for FACS analysis.
2.11. In Vivo Cellular Uptake of Cy5-siRNA-Loaded NIR-Labelled NonNP
To quantify the in vivo cellular uptake of Cy5-labeled scsiRNA complexed with NIR-labeled NonNP via FACS analysis, isolated liver cells obtained from mice injected with 2mg/kg scsiRNA/NonNP were stained with fluorescent-labeled antibodies. The following antibodies were applied according the manufacture’s protocol for intra/extracellular staining: macrophages: F4/80-PE (clone BM8) or CD11b-BV421 (clone M1/70, both from eBiolegend, San Diego, CA, USA); hepatocytes: albumin-PE (Clone #188835, R&D, Boston, MA, USA) and endothelial cells; CD31-PE (MEC13.3, eBiolegend, San Diego, CA, USA); granulocytes: Gr1+ (RB6-8C5, MEC13.3, eBiolegend, San Diego, CA, USA); NKT-cells: CD4-FITC (GK1.5) and NK1.1 (PK136, both from eBiolegend, San Diego, USA); dendritic cells: F4/80-PE (clone BM8) and CD11c-BV421 (clone N418, both from eBiolegend, San Diego, CA, USA); macrophages: M2 phenotype CD45-FITC (clone 30F11) combined with F4/80-PE (clone BM8) and CD206-BV421 (C068C2, all from eBiolegend, San Diego, CA, USA). Moreover, eFluor 506 (eBioscience, San Diego, CA, USA) was used for live/dead staining. Afterwards, cells were fixed with 4% formaldehyde/PBS for 15 min at 37 °C and analyzed by a BD FACS Canto II (BD Bioscience, Canada, Mississauga) Flow cytometer. 10,000–50,000 cells were measured per staining. The obtained data were analyzed by Flowing Software 2.5.0 (Perttu Terho, Turku Centre for Biotechnology, Finland).
2.12. Confocal Laser Scanning Microscopy of CD206 Immunofluorescent Stained Liver Cryosections from Mice Treated with Cy5-siRNA-Loaded NIR-(Non-)ManNP
Directly after sacrifice, parts of the middle and right liver lobe from mice treated with 2 mg/kg scsiRNA/(Non-)ManNP were embedded in OCT compound from Sakura (Staufen, Germany) and stored at −20 °C. Liver cryosections were cut at 6 µm with a Leica CM 1950 cryostat (Wetzlar, Germany) and mounted on superfrost ultra plus adhesion histological slides from Thermo Fisher Scientific (Schwerte, Germany). Slides were stored at −70 °C for subsequent staining. Cryo slides were rehydrated with PBS at room temperature for 20 min, fixed with fixation buffer (intracellular fixation&permeabilization buffer set, eBioscience, Frankfurt, Germany) for 10 min and washed three times with a permeabilization buffer. Tissues were surrounded with a hydrophobic barrier using a Dako pen from Agilent (Santa Clara, CA, USA). Afterwards, sections were blocked with 5% donkey serum in a permeabilization buffer for 1 h at room temperature and incubated overnight at 4 °C with CD206-FITC antibodies (C068C2, eBioscience, San Diego, CA, USA) diluted 1:100 in permeabilization buffer. After 12 h, slides were washed with a permeabilization buffer and H2O, covered with coverslips after prior application of antifade mounting medium, and stained with DAPI from VECTASHIELD (Orton Southgate, UK).
4. Conclusions
In this study α-mannosylated siRNA nanohydrogel carriers were evaluated for specific targeting of profibrotic M2 polarized macrophages through the overexpressed mannose receptor CD206 in liver fibrotic mice. Carriers were synthesized and characterized as described by us in detail previously, yielding chemically well-defined nanohydrogel particles that carry a high degree of α-mannosyl functionalization on their surface [
23].
To qualify for in vivo application, siRNA carriers had to meet three primary requirements of in vitro screening. First, cytotoxic or pro-inflammatory effects of particles had to be ruled out at concentrations relevant for in vivo therapy. Here, ManNP were tested in the human macrophage cell line THP-1 that mirrors characteristics of liver resident macrophages. Equal to murine cells, α-mannosyl functionalization did not compromise THP-1 cell viability at reasonable concentrations (25–50 nM siRNA). ManNP were even better tolerated by THP-1 cells up to very high siRNA concentrations (>320 nM) than their non-functionalized counterparts. Secondly, carriers had to demonstrate preferred uptake in disease relevant cells. ManNP showed a dose-dependent uptake by cells relevant for fibrogenesis. However, in comparison to NonNP, unspecific cellular uptake in liver epithelial cells and fibroblasts was lower for mannose functionalized carriers, which may be due to the mannose corona that prevents unspecific uptake in non-targeted cells. This will be an advantage over non-functionalized NPs, reducing, e.g., unwanted off-target side effects. Thus, relative uptake of ManNP was higher in M2 polarized macrophages, overexpressing the mannose receptor CD206, than in other cell types, as demonstrated by us before [
23]. Third, knockdown efficiency of the siRNA-loaded carriers needs to be robust for the target transcript in relevant cells. Here, anti-
CSFR1 siRNA-loaded ManNP have already been demonstrated to induce solid knockdown in M2 polarized primary macrophages, while the (low CD206 expressing) M1 phenotype was not affected [
23]. Taken together, our ManNP met all these key criteria for their application in vivo.
In the current in vivo liver fibrosis model enriched in M2 macrophages, fluorescently labeled ManNP loaded with Cy5-scsiRNA were rapidly sequestrated in the fibrotic liver after intravenous administration comparable to their non-functionalized analogue. In vivo biodistribution of the carriers was independent from fibrosis as accumulation of the complexes was also observed in healthy mice. Since the carriers also accumulate in livers of healthy mice, in vivo siRNA delivery to M2 macrophages appears feasible even at the early stages of chronic liver disease, allowing preventive treatment.
In these in vivo studies, we also looked at the cellular uptake of both ManNP and NonNP in those fibrotic livers. As predicted by the previous in vitro studies, cellular uptake of ManNP in M2-type macrophages was higher than in other nonparenchymal, i.e., fibroblastic and endothelial liver cells, and was also increased compared to their non-functionalized counterparts, while particles without mannose seem to be randomly internalized independent from the (macrophage) phenotype.
In summary, the introduced mannose surface functionalization prevented both unspecific uptake in non-target cells and significantly enhanced uptake in M2 polarized macrophages with a high expression of CD206 in vivo. Moreover, the introduced functionalization did not compromise the good biocompatibility of the nanohydrogel particles. Finally, in view of effective siRNA delivery by the non-functionalized NP in vivo [
25,
26], our ManNP qualify as improved nanocarriers for M2 macrophage-specific siRNA delivery. As the next generation of nanohydrogel particles, ManNP represent a promising siRNA carrier platform for further translational therapeutic studies for repolarization of immunosuppressive and profibrotic M2-type macrophages.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}