Halobacterium salinarum virus ChaoS9, a Novel Halovirus Related to PhiH1 and PhiCh1

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Host Strain, Virus Isolation, Cultivation, and Purification
2.2. DNA Sequencing and Assembly of the ChaoS9 Genome
2.3. DNA Resequencing of Natrialba Virus phiCh1
2.4. Electron Microscopy of Virus
2.5. Protein Analyses of Purified Virus
2.6. Bioinformatics Analyses
Data Availability
3. Results
3.1. Isolation of Halovirus ChaoS9
3.2. Virus Proteins
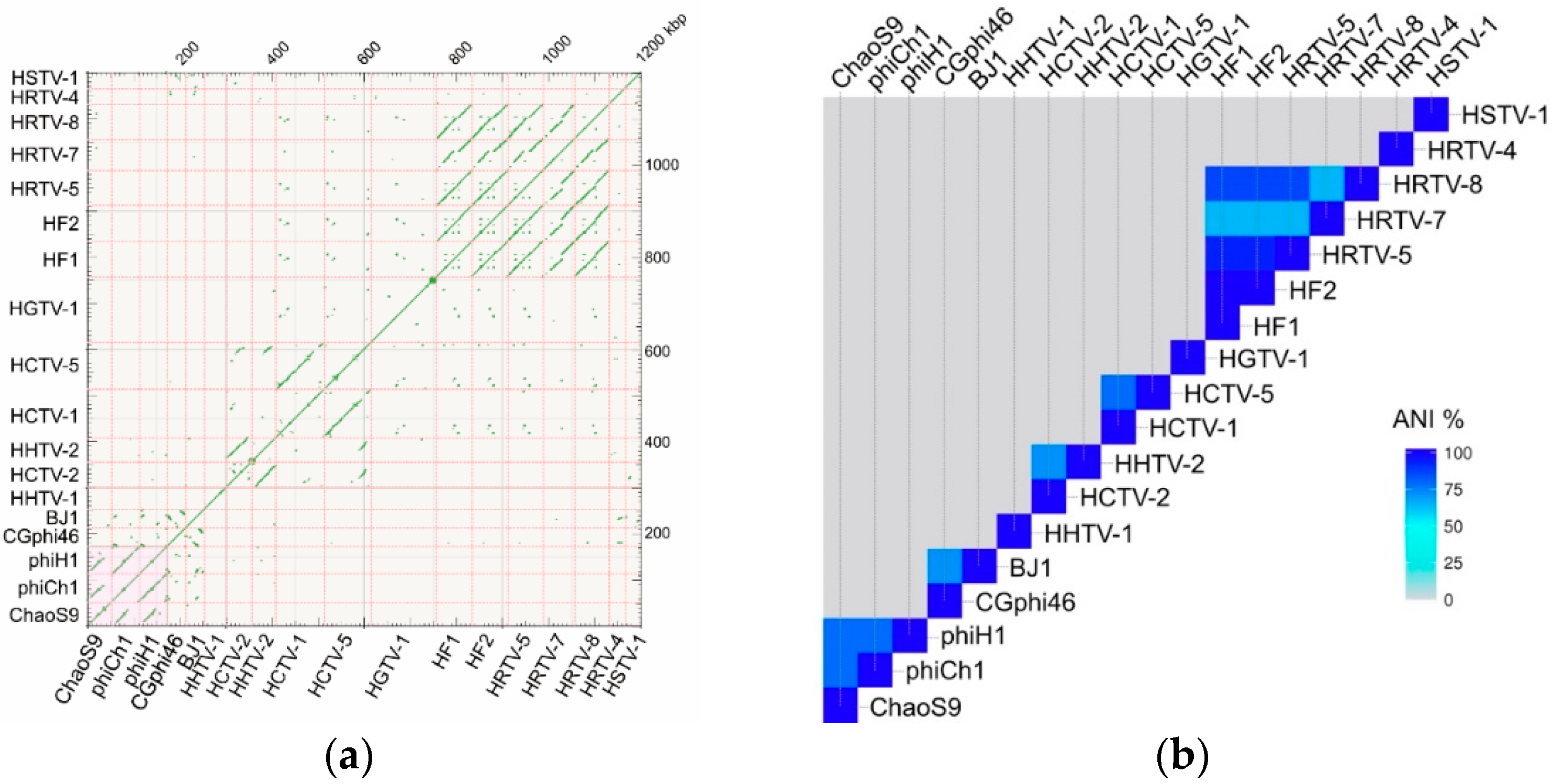
3.3. ChaoS9 Genome and Sequence
3.4. Resequencing the Genome of Halovirus PhiCh1
3.5. Organisation of the ChaoS9 Genome
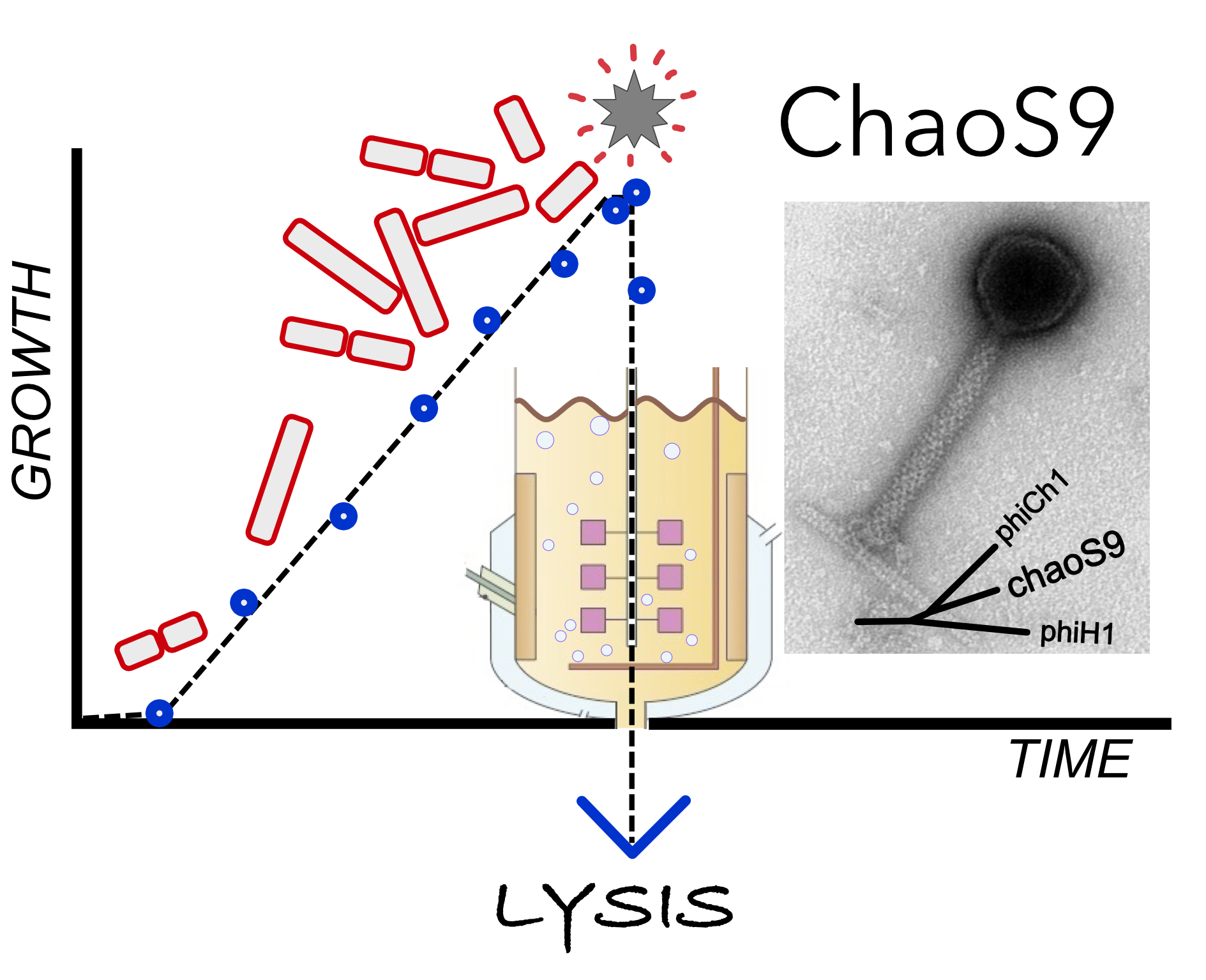
3.6. Related Provirus-Like Matches in Haloarchaea
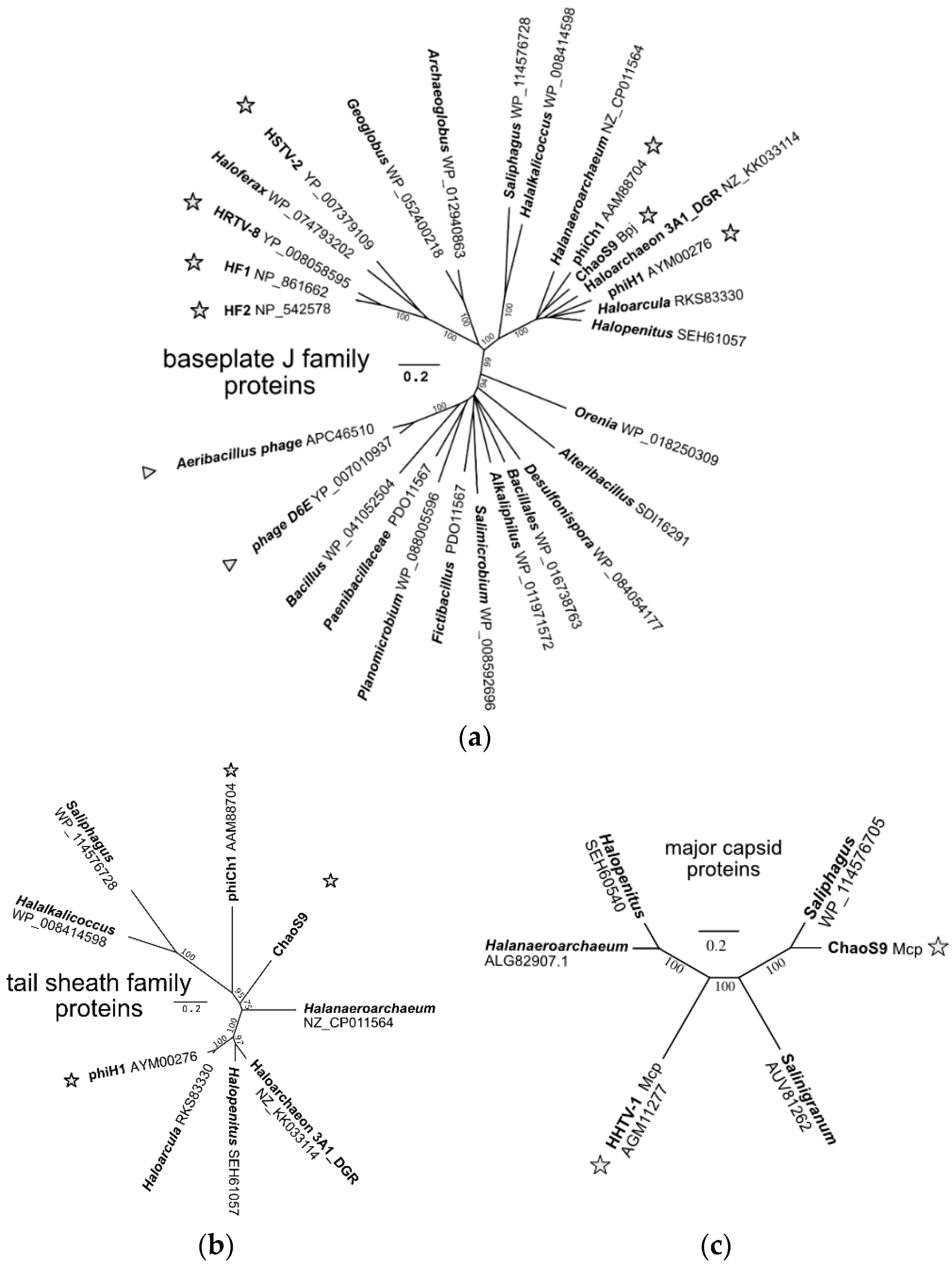
3.7. A Diverse Family of Haloviruses
3.8. CRISPR Spacer Matches to ChaoS9
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dyall-Smith, M.; Tang, S.L.; Bath, C. Haloarchaeal viruses: How diverse are they? Res. Microbiol. 2003, 154, 309–313. [Google Scholar] [CrossRef]
- Krupovic, M.; Cvirkaite-Krupovic, V.; Iranzo, J.; Prangishvili, D.; Koonin, E.V. Viruses of archaea: Structural, functional, environmental and evolutionary genomics. Virus Res. 2017, 244, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Bratbak, G.; Heldal, M. Occurrence of virus-like particles in the Dead Sea. Extremophiles 1997, 1, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P.; Bamford, D.H. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J. Mol. Biol. 2010, 397, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Sencilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.C.; Bowman, C.A.; Atanasova, N.S.; Osterlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 2013, 10, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Sencilo, A.; Roine, E. A glimpse of the genomic diversity of haloarchaeal tailed viruses. Front. Microbiol. 2014, 5, 84. [Google Scholar] [PubMed]
- Atanasova, N.S.; Bamford, D.H.; Oksanen, H.M. Virus-host interplay in high salt environments. Environ. Microbiol. Rep. 2016, 8, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, Y.; Wang, S.; Yang, D.; Cheng, Y.; Hu, J.; Chen, J.; Mei, Y.; Shen, P.; Bamford, D.H.; et al. Temperate membrane-containing halophilic archaeal virus SNJ1 has a circular dsDNA genome identical to that of plasmid pHH205. Virology 2012, 434, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Liu, Y.; Wang, Y.; Zhang, Z.; Oksanen, H.M.; Bamford, D.H.; Chen, X. Identification and characterization of SNJ2, the first temperate pleolipovirus integrating into the genome of the SNJ1-lysogenic archaeal strain. Mol. Microbiol. 2015, 98, 1002–1020. [Google Scholar] [CrossRef] [PubMed]
- Demina, T.A.; Pietila, M.K.; Svirskaite, J.; Ravantti, J.J.; Atanasova, N.S.; Bamford, D.H.; Oksanen, H.M. Archaeal Haloarcula californiae Icosahedral virus 1 highlights conserved elements in icosahedral membrane-containing DNA viruses from extreme environments. MBio 2016, 7, e00699-16. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, R.; Oesterhelt, D. Bacteriorhodopsin-mediated photophosphorylation in Halobacterium halobium. Eur. J. Biochem. 1977, 77, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Michel, H.; Oesterhelt, D. Electrochemical proton gradient across the cell membrane of Halobacterium halobium: Effect of N,N’-dicyclohexylcarbodiimide, relation to intracellular adenosine triphosphate, adenosine diphosphate, and phosphate concentration, and influence of the potassium gradient. Biochemistry 1980, 19, 4607–4614. [Google Scholar] [PubMed]
- Haupts, U.; Tittor, J.; Oesterhelt, D. Closing in on bacteriorhodopsin: Progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 367–399. [Google Scholar] [CrossRef] [PubMed]
- Mirfeizollahi, A.; Yakhchali, B.; Deldar, A.A.; Karkhane, A.A. In silico and experimental improvement of bacteriorhodopsin production in Halobacterium salinarum R1 by increasing DNA-binding affinity of Bat through Q661R/Q665R substitutions in HTH motif. Extremophiles 2019, 23, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Stoeckenius, W.; Lozier, R.H.; Bogomolni, R.A. Bacteriorhodopsin and the purple membrane of halobacteria. Biochim. Biophys. Acta (BBA)—Rev. Bioenerg. 1979, 505, 215–278. [Google Scholar] [CrossRef]
- Baliga, N.S.; DasSarma, S. Saturation mutagenesis of the TATA box and upstream activator sequence in the haloarchaeal bop gene promoter. J. Bacteriol. 1999, 181, 2513–2518. [Google Scholar] [PubMed]
- Baliga, N.S.; Kennedy, S.P.; Ng, W.V.; Hood, L.; DasSarma, S. Genomic and genetic dissection of an archaeal regulon. Proc. Natl. Acad. Sci. USA 2001, 98, 2521–2525. [Google Scholar] [CrossRef] [PubMed]
- Baliga, N.S.; Pan, M.; Goo, Y.A.; Yi, E.C.; Goodlett, D.R.; Dimitrov, K.; Shannon, P.; Aebersold, R.; Ng, W.V.; Hood, L. Coordinate regulation of energy transduction modules in Halobacterium sp. analyzed by a global systems approach. Proc. Natl. Acad. Sci. USA 2002, 99, 14913–14918. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Kim, J.M.; Molinari, E.; DasSarma, S. Genetic and topological analyses of the bop promoter of Halobacterium halobium: Stimulation by DNA supercoiling and non-B-DNA structure. J. Bacteriol. 1996, 178, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; Nordmann, B.; Storch, K.F.; Gruenberg, H.; Rodewald, K.; Oesterhelt, D. A family of halobacterial transducer proteins. FEMS Microbiol. Lett. 1996, 139, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Storch, K.F.; Rudolph, J.; Oesterhelt, D. Car: A cytoplasmic sensor responsible for arginine chemotaxis in the archaeon Halobacterium salinarum. EMBO J. 1999, 18, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.B.; Moineau, S.; Quiberoni, A. Bacteriophages and dairy fermentations. Bacteriophage 2012, 2, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H.; Zillig, W.; Pfaffle, M.; Schnabel, R.; Michel, H.; Delius, H. Halobacterium halobium phage ΦH. EMBO J. 1982, 1, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Soppa, J.; Oesterhelt, D. Bacteriorhodopsin mutants of Halobacterium sp. GRB. I. The 5-bromo-2′-deoxyuridine selection as a method to isolate point mutants in halobacteria. J. Biol. Chem. 1989, 264, 13043–13048. [Google Scholar] [PubMed]
- Gordon, D. Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinform. 2003, 2, 11.2.1–11.2.43. [Google Scholar]
- Dyall-Smith, M.; Pfeifer, F.; Witte, A.; Oesterhelt, D.; Pfeiffer, F. Complete genome sequence of the model halovirus phiH1 (ΦH1). Genes 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Lomsadze, A.; Gemayel, K.; Tang, S.; Borodovsky, M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018, 28, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Siddaramappa, S.; Challacombe, J.F.; Decastro, R.E.; Pfeiffer, F.; Sastre, D.E.; Gimenez, M.I.; Paggi, R.A.; Detter, J.C.; Davenport, K.W.; Goodwin, L.A.; et al. A comparative genomics perspective on the genetic content of the alkaliphilic haloarchaeon Natrialba magadii ATCC 43099T. BMC Genom. 2012, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guerois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, I.; Loots, G.G.; Hardison, R.C.; Miller, W.; Stubbs, L. zPicture: Dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res. 2004, 14, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRcompar: A website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2008, 36, W145–W148. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Chen, I.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 2017, 45, D457–D465. [Google Scholar] [CrossRef] [PubMed]
- Skennerton, C.T.; Imelfort, M.; Tyson, G.W. Crass: Identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 2013, 41, e105. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhu, Q.; Huang, D.; Zhao, S.; Jan Lo, L.; Peng, J. An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Sci. Rep. 2015, 5, 13370. [Google Scholar] [CrossRef] [PubMed]
- ICTV Report. ICTV Online (10th) Report on Virus Taxonomy. Available online: https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=20170459 (accessed on 19 March 2018).
- Schnabel, H. An immune strain of Halobacterium halobium carries the invertible L segment of phage ΦH as a plasmid. Proc. Natl. Acad. Sci. USA 1984, 81, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.; Baranyi, U.; Klein, R.; Sulzner, M.; Luo, C.; Wanner, G.; Krüger, D.H.; Lubitz, W. Characterization of Natronobacterium magadii phage ϕCh1, a unique archaeal phage containing DNA and RNA. Mol. Microbiol. 1997, 23, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Baranyi, U.; Rössler, N.; Greineder, B.; Scholz, H.; Witte, A. Natrialba magadii virus ϕCh1: First complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol. Microbiol. 2002, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Gene regulation in halophage ΦH; more than promoters. Syst. Appl. Microbiol. 1993, 16, 591–596. [Google Scholar] [CrossRef]
- VIRFAM. Remote Homology Detection of Viral Protein Families. Available online: http://biodev.cea.fr/virfam/ (accessed on 8 October 2018).
- Klein, R.; Rossler, N.; Iro, M.; Scholz, H.; Witte, A. Haloarchaeal myovirus phiCh1 harbours a phase variation system for the production of protein variants with distinct cell surface adhesion specificities. Mol. Microbiol. 2012, 83, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H.; Zillig, W. Circular structure of the genome of phage ΦH in a lysogenic Halobacterium halobium. Mol. Gen. Genet. 1984, 193, 422–426. [Google Scholar] [CrossRef]
- Iro, M.; Klein, R.; Galos, B.; Baranyi, U.; Rossler, N.; Witte, A. The lysogenic region of virus phiCh1: Identification of a repressor-operator system and determination of its activity in halophilic Archaea. Extremophiles 2007, 11, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Gropp, F.; Grampp, B.; Stolt, P.; Palm, P.; Zillig, W. The immunity-conferring plasmid pϕHL from the Halobacterium salinarium phage ϕH: Nucleotide sequence and transcription. Virology 1992, 190, 45–54. [Google Scholar] [CrossRef]
- Ken, R.; Hackett, N.R. Halobacterium halobium strains lysogenic for phage phiH contain a protein resembling coliphage repressors. J. Bacteriol. 1991, 173, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo studies on the effects of immunity genes on early lytic transcription in the Halobacterium salinarium phage ϕH. Mol. Gen. Genet. 1992, 235, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Transcription of the halophage ΦH repressor gene is abolished by transcription from an inversely oriented lytic promoter. FEBS Lett. 1994, 344, 125–128. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Schuster, S.C.; Broicher, A.; Falb, M.; Palm, P.; Rodewald, K.; Ruepp, A.; Soppa, J.; Tittor, J.; Oesterhelt, D. Evolution in the laboratory: The genome of Halobacterium salinarum strain R1 compared to that of strain NRC-1. Genomics 2008, 91, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Svirskaite, J.; Oksanen, H.M.; Daugelavicius, R.; Bamford, D.H. Monitoring physiological changes in haloarchaeal cell during virus release. Viruses 2016, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Mavrich, T.N.; Garlena, R.A.; Guerrero-Bustamante, C.A.; Jacobs-Sera, D.; Montgomery, M.T.; Russell, D.A.; Warner, M.H.; Science Education Alliance-Phage Hunters Advancing, G.; Evolutionary, S.; et al. Bacteriophages of Gordonia spp. display a spectrum of diversity and genetic relationships. MBio 2017, 8, e01069-17. [Google Scholar] [CrossRef] [PubMed]
- Summer, E.J.; Enderle, C.J.; Ahern, S.J.; Gill, J.J.; Torres, C.P.; Appel, D.N.; Black, M.C.; Young, R.; Gonzalez, C.F. Genomic and biological analysis of phage Xfas53 and related prophages of Xylella fastidiosa. J. Bacteriol. 2010, 192, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Smith, M.C.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Samson, J.E.; Moineau, S. Bacteriophages in food fermentations: New frontiers in a continuous arms race. Annu. Rev. Food Sci. Technol. 2013, 4, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2014, 468–470, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Common evolutionary origin of procapsid proteases, phage tail tubes, and tubes of bacterial type VI secretion systems. Structure 2016, 24, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Grose, J.H. Contributions of P2- and P22-like prophages to understanding the enormous diversity and abundance of tailed bacteriophages. Virology 2016, 496, 255–276. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus 1 | Head Diameter (nm) | Tail Length × Width (nm) | Morphotype | Plaque Morphology | Unit Genome Length 2 (nt) | %G + C | GenomeEnds in Virus 3 | Temperate (Genome Form) |
|---|---|---|---|---|---|---|---|---|
| ChaoS9 | 61 | 128 × 17 | myovirus | turbid | 55,145 | 65.3 | ds, linear, TR, CP, >1 unit length | ? |
| phiH1 | 64 | 170 × 18 | myovirus | turbid | 58,072 | 63.7 | ds, linear, TR, CP, >1 unit length | Yes, provirus is a plasmid (circular, ds, 1 unit length) |
| phiCh1 | 70 | 130 × 20 | myovirus | turbid | 58,487 | 61.9 | ds, linear, TR, CP, >1 unit length | Yes, provirus is a plasmid (circular, ds, 1 unit length) |
| Start (nt) | Stop (nt) | Locus_Tag | Length (bp) | Direction | Gene | Product | Homologs 1: phiCh1, pNMAG03 | Homologs 2: phiH1, [Other] |
|---|---|---|---|---|---|---|---|---|
| 100 | 669 | ChaoS9_005 | 570 | + | - | HTH domain protein | PhiCh1_005, PhiCh1p02, ORF1, Nmag_4251 | PhiH1_005 |
| 656 | 2302 | ChaoS9_010 | 1647 | + | terL | terminase large subunit TerL | - | [HALG_00007] |
| 2316 | 3944 | ChaoS9_015 | 1629 | + | por | portal protein Por | - | [HGTV1_7] |
| 3937 | 4083 | ChaoS9_020 | 147 | + | - | CxxC motif protein | - | ORPHAN |
| 4086 | 5273 | ChaoS9_025 | 1188 | + | - | putative phage head assembly protein, SPP1_gp7 family | - | [C478_10461] |
| 5384 | 7300 | ChaoS9_030 | 1917 | + | - | probable prohead protease protein | - | [HLASA_2034] |
| 7303 | 7755 | ChaoS9_035 | 453 | + | - | uncharacterized protein | - | [HLASA_2033] |
| 7801 | 8928 | ChaoS9_040 | 1128 | + | - | major capsid protein MCP | - | 3 [HLASA_2032; HHTV1_21] |
| 8944 | 9363 | ChaoS9_045 | 420 | + | - | uncharacterized protein | PhiCh1_055, PhiCh1p13, ORF12, Nmag_4261 | 6 PhiH1_050, [HLASA_2031] |
| 9398 | 9784 | ChaoS9_050 | 387 | + | - | uncharacterized protein | - | [HLASA_2030] |
| 9781 | 10191 | ChaoS9_055 | 411 | + | hco | head closure protein Hco, type 1 | PhiCh1_065, PhiCh1p15, ORF14, Nmag_4263 | 5 PhiH1_060, [WP_054519912] |
| 10188 | 10421 | ChaoS9_060 | 234 | + | - | uncharacterized protein | - | ORPHAN |
| 10414 | 10701 | ChaoS9_065 | 288 | + | - | uncharacterized protein | PhiCh1_070, PhiCh1p16, ORF15, Nmag_4264 | 6 PhiH1_065, [HLASA_2029] |
| 10703 | 11149 | ChaoS9_070 | 447 | + | nep | putative neck protein Nep, type 1 | PhiCh1_075, PhiCh1p17, ORF16, Nmag_4265 | PhiH1_070 [HLASA_2028] |
| 11156 | 11746 | ChaoS9_075 | 591 | + | tco | tail completion protein Tco, type 1 | PhiCh1_080, PhiCh1p18, ORF17, Nmag_4266 | PhiH1_075 [HLASA_2027] |
| 11767 | 13071 | ChaoS9_080 | 1305 | + | - | tail sheath protein | PhiCh1_085, PhiCh1p19, ORF18, Nmag_4267 | PhiH1_080 [HLASA_2026] |
| 13082 | 13480 | ChaoS9_085 | 399 | + | - | predicted tail tube protein | PhiCh1_090, PhiCh1p20, ORF19, Nmag_4268 | PhiH1_085 [WP_054519907] |
| 13492 | 13932 | ChaoS9_090 | 441 | + | - | uncharacterized protein | PhiCh1_095, PhiCh1p21, ORF20, Nmag_4269 | PhiH1_090 [HLASA_2025] |
| 13935 | 14144 | ChaoS9_095 | 210 | + | - | uncharacterized protein | - | [WP_054519905] |
| 14147 | 16891 | ChaoS9_100 | 2745 | + | tpm | tape-measure tail protein Tpm | 7 PhiCh1_105, PhiCh1p23+PhiCh1p24, ORF22+ORF23, Nmag_4272 | PhiH1_100 [HLASA_2024] |
| 16895 | 17422 | ChaoS9_105 | 528 | + | - | uncharacterized protein | PhiCh1_110, PhiCh1p25, ORF24, Nmag_4273 | PhiH1_105 [HLASA_2023] |
| 17423 | 17767 | ChaoS9_110 | 345 | + | - | uncharacterized protein | PhiCh1_115, PhiCh1p26, ORF25, Nmag_4274 | PhiH1_110 [HLASA_2022] |
| 17771 | 18604 | ChaoS9_115 | 834 | + | - | uncharacterized protein | 7 PhiCh1_120, PhiCh1p27+PhiCh1p28, ORF26+ORF27, Nmag_4275 | PhiH1_115 [HLASA_2021] |
| 18644 | 18787 | ChaoS9_120 | 144 | + | - | CxxC motif protein | - | PhiH1_120 |
| 18784 | 19335 | ChaoS9_125 | 552 | + | - | uncharacterized protein | PhiCh1_125, PhiCh1p29, ORF28, Nmag_4276 | PhiH1_125 [HLASA_2020] |
| 19338 | 19700 | ChaoS9_130 | 363 | + | - | virus-related protein | - | PhiH1_135 |
| 19697 | 20062 | ChaoS9_135 | 366 | + | - | uncharacterized protein | PhiCh1_130, PhiCh1p30, ORF29, Nmag_4277 | PhiH1_140 [HLASA_2019] |
| 20069 | 21328 | ChaoS9_140 | 1260 | + | bpj | baseplate J family protein Bpj | PhiCh1_135, PhiCh1p31, ORF30, Nmag_4278 | PhiH1_145 [HLASA_2018] |
| 21321 | 21929 | ChaoS9_145 | 609 | + | - | uncharacterized protein | PhiCh1_140, PhiCh1p32, ORF31, Nmag_4279 | PhiH1_150 [HLASA_2017] |
| 21933 | 22517 | ChaoS9_150 | 585 | + | - | uncharacterized protein | PhiCh1_145, PhiCh1p33, ORF32, Nmag_4280 | - [HLASA_2016] |
| 22514 | 23122 | ChaoS9_155 | 609 | + | - | uncharacterized protein | PhiCh1_150, PhiCh1p34, ORF33, Nmag_4281 | - [HLASA_2015] |
| 23115 | 24698 | ChaoS9_160 | 1584 | + | - | repeat-containing tail fiber protein | 8 PhiCh1_155+ PhiCh1_175, PhiCh1p35+PhiCh1p37, ORF34+ORF36, Nmag_4282+Nmag_4286 | PhiH1_165+ PhiH1_185 |
| 24702 | 24986 | ChaoS9_165 | 285 | + | - | uncharacterized protein | 8 PhiCh1_160+ PhiCh1_170, Nmag_4285+Nmag_4283 | PhiH1_180+ PhiH1_170 |
| 25024 | 25698 | ChaoS9_170 | 675 | + | int1 | tyrosine integrase/recombinase Int1 | PhiCh1_165, PhiCh1p36, ORF35, Nmag_4284 | PhiH1_175 |
| 25709 | 25996 | ChaoS9_175 | 288 | - | - | uncharacterized protein | 8 PhiCh1_160+ PhiCh1_170, Nmag_4285+Nmag_4283 | PhiH1_180+ PhiH1_170 |
| 25999 | 26145 | ChaoS9_180 | 147 | - | - | repeat-containing tail fiber protein (C-term) (nonfunctional)9 | * 9 | * 9 |
| 26199 | 27455 | ChaoS9_185 | 1257 | - | tnpB | IS1341-type transposase TnpB | - | PhiH1_340 |
| 27457 | 27849 | ChaoS9_190 | 393 | - | tnpA | IS200-type transposase TnpA | - | PhiH1_335 |
| 27906 | 28820 | ChaoS9_195 | 915 | - | - | repeat-containing tail fiber protein (N-term) (nonfunctional) | 8 PhiCh1_155+ PhiCh1_175, PhiCh1p35+PhiCh1p37, ORF34+ORF36, Nmag_4282+Nmag_4286 | PhiH1_165+ PhiH1_185 |
| 28854 | 29579 | ChaoS9_200 | 726 | + | - | transmembrane domain protein | PhiCh1_180, PhiCh1p38, ORF37, Nmag_4287 | - |
| 29589 | 29861 | ChaoS9_205 | 273 | - | - | HTH domain protein | PhiCh1_185, PhiCh1p39, ORF38, Nmag_4288 | - |
| 29933 | 30241 | ChaoS9_210 | 309 | - | - | uncharacterized protein | PhiCh1_190, PhiCh1p40, ORF39, Nmag_4289 | PhiH1_220 |
| 30238 | 30813 | ChaoS9_215 | 576 | - | - | glutamine amidotransferase domain protein, class-II | PhiCh1_195, PhiCh1p41, ORF40, Nmag_4290 | - |
| 30818 | 31891 | ChaoS9_220 | 1074 | - | - | uncharacterized protein | 7 PhiCh1_200, PhiCh1p42+PhiCh1p43, ORF41+ORF42, Nmag_4291 | - |
| 32030 | 32266 | ChaoS9_225 | 237 | + | - | uncharacterized protein | - | ORPHAN |
| 32339 | 32584 | ChaoS9_230 | 246 | + | - | uncharacterized protein | - | PhiH1_225 |
| 32581 | 33009 | ChaoS9_235 | 429 | + | - | VapC family toxin | - | [BRC75_08225] |
| 33098 | 33448 | ChaoS9_240 | 351 | + | - | uncharacterized protein | PhiCh1_230, Nmag_4297 | PhiH1_250 |
| 33457 | 34059 | ChaoS9_245 | 603 | - | int2 | tyrosine integrase/recombinase Int2 | PhiCh1_215, PhiCh1p46, ORF45, Nmag_4294 | PhiH1_240 |
| 34241 | 34432 | ChaoS9_250 | 192 | - | - | uncharacterized protein | - | ORPHAN |
| 34507 | 35055 | ChaoS9_255 | 549 | - | - | uncharacterized protein | - | PhiH1_255 [C466_00612] |
| 35048 | 35902 | ChaoS9_260 | 855 | - | - | Plasmid partition protein ParA | PhiCh1_220, PhiCh1p47, ORF46, Nmag_4295 | PhiH1_265 |
| 35976 | 36440 | ChaoS9_265 | 465 | - | - | transmembrane domain protein | - | PhiH1_210 |
| 36460 | 38241 | ChaoS9_270 | 1782 | - | - | uncharacterized protein | - | [AV929_12240] |
| 38243 | 38857 | ChaoS9_275 | 615 | - | - | uncharacterized protein | - | [CRI94_04435] |
| 38850 | 39080 | ChaoS9_280 | 231 | - | - | CxxC motif protein | - | [HALLA_11930] |
| 39073 | 39240 | ChaoS9_285 | 168 | - | - | transmembrane domain protein | - | ORPHAN |
| 39233 | 40492 | ChaoS9_290 | 1260 | - | - | uncharacterized protein | - | [DM826_07215] |
| 40485 | 40673 | ChaoS9_295 | 189 | - | - | CxxC motif protein | - | ORPHAN |
| 40663 | 41181 | ChaoS9_300 | 519 | - | - | uncharacterized protein | - | [DJ71_18565] |
| 41174 | 41611 | ChaoS9_305 | 438 | - | - | HNH-type endonuclease/MarR family transcription regulator | - | 4 [DJ70_12900; B4589_07635] |
| 41613 | 41870 | ChaoS9_310 | 258 | - | - | HTH domain protein | - | [Natpe_3999] |
| 41997 | 42602 | ChaoS9_315 | 606 | + | - | uncharacterized protein | - | [C480_10020] |
| 42605 | 43051 | ChaoS9_320 | 447 | + | - | uncharacterized protein | - | [Natgr_3468] |
| 43044 | 43250 | ChaoS9_325 | 207 | + | - | uncharacterized protein | - | ORPHAN |
| 43250 | 43621 | ChaoS9_330 | 372 | + | - | uncharacterized protein | - | [OSG_eHP13_00215] |
| 43621 | 44127 | ChaoS9_335 | 507 | + | - | uncharacterized protein | - | ORPHAN |
| 44124 | 44255 | ChaoS9_340 | 132 | + | - | CxxC motif protein | - | [SAMN04488133_0114] |
| 44342 | 45400 | ChaoS9_345 | 1059 | + | orc1 | Orc1-type DNA replication protein | - | [HLASA_2006] |
| 45482 | 45709 | ChaoS9_350 | 228 | - | - | uncharacterized protein | - | ORPHAN |
| 45837 | 45992 | ChaoS9_355 | 156 | + | - | uncharacterized protein | - | [HALDL1_16575] |
| 46369 | 46905 | ChaoS9_360 | 537 | + | - | uncharacterized protein | PhiCh1_295, PhiCh1p65, ORF64, Nmag_4216 | 5 [C472_00499] |
| 46902 | 47177 | ChaoS9_365 | 276 | + | - | uncharacterized protein | - | ORPHAN |
| 47179 | 47979 | ChaoS9_370 | 801 | + | - | zinc-finger domain protein | - | [DJ84_18225] |
| 47976 | 48068 | ChaoS9_375 | 93 | + | - | uncharacterized protein | - | ORPHAN |
| 48065 | 48397 | ChaoS9_380 | 333 | + | - | uncharacterized protein | - | ORPHAN |
| 48394 | 51690 | ChaoS9_385 | 3297 | + | repH | plasmid replication protein RepH | PhiCh1_245, PhiCh1p55, ORF54, Nmag_4299 | PhiH1_285 |
| 51683 | 51925 | ChaoS9_390 | 243 | + | - | MarR family transcription regulator | - | [AV929_12115] |
| 51932 | 52591 | ChaoS9_395 | 660 | + | - | CxxC motif protein | - | [C443_17983] |
| 53208 | 53453 | ChaoS9_400 | 246 | + | - | transmembrane domain protein | PhiCh1_440, PhiCh1p93, ORF92, Nmag_4244 | PhiH1_460 |
| 53446 | 53769 | ChaoS9_405 | 324 | + | - | transmembrane domain protein | PhiCh1_445, PhiCh1p94, ORF93, Nmag_4245 | 10 PhiH1_465 |
| 53766 | 54455 | ChaoS9_410 | 690 | + | - | uncharacterized protein | - | 3 [halTADL_2427; HGTV1_34] |
| 54460 | 54705 | ChaoS9_415 | 246 | + | - | DUF217 domain protein | PhiCh1_460, PhiCh1p97, ORF96, Nmag_4248 | - |
| 54734 | 54922 | ChaoS9_420 | 189 | + | - | CxxC motif protein | PhiCh1_465, PhiCh1p98, ORF97, Nmag_4249 | PhiH1_480 |
| 54915> | <63 | ChaoS9_425 | 294 | + | terS | terminase small subunit TerS | PhiCh1_470, PhiCh1p01, ORF98, Nmag_4250 | PhiH1_485 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyall-Smith, M.; Palm, P.; Wanner, G.; Witte, A.; Oesterhelt, D.; Pfeiffer, F. Halobacterium salinarum virus ChaoS9, a Novel Halovirus Related to PhiH1 and PhiCh1. Genes 2019, 10, 194. https://doi.org/10.3390/genes10030194
Dyall-Smith M, Palm P, Wanner G, Witte A, Oesterhelt D, Pfeiffer F. Halobacterium salinarum virus ChaoS9, a Novel Halovirus Related to PhiH1 and PhiCh1. Genes. 2019; 10(3):194. https://doi.org/10.3390/genes10030194
Chicago/Turabian StyleDyall-Smith, Mike, Peter Palm, Gerhard Wanner, Angela Witte, Dieter Oesterhelt, and Friedhelm Pfeiffer. 2019. "Halobacterium salinarum virus ChaoS9, a Novel Halovirus Related to PhiH1 and PhiCh1" Genes 10, no. 3: 194. https://doi.org/10.3390/genes10030194
APA StyleDyall-Smith, M., Palm, P., Wanner, G., Witte, A., Oesterhelt, D., & Pfeiffer, F. (2019). Halobacterium salinarum virus ChaoS9, a Novel Halovirus Related to PhiH1 and PhiCh1. Genes, 10(3), 194. https://doi.org/10.3390/genes10030194