Cohesin-Mediated Genome Architecture Does Not Define DNA Replication Timing Domains



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Sorting
2.3. Synchronization
2.4. Flow Cytometry
2.5. RepliSeq
2.6. Immunofluorescence
2.7. RepliSeq Computational Analysis
3. Results
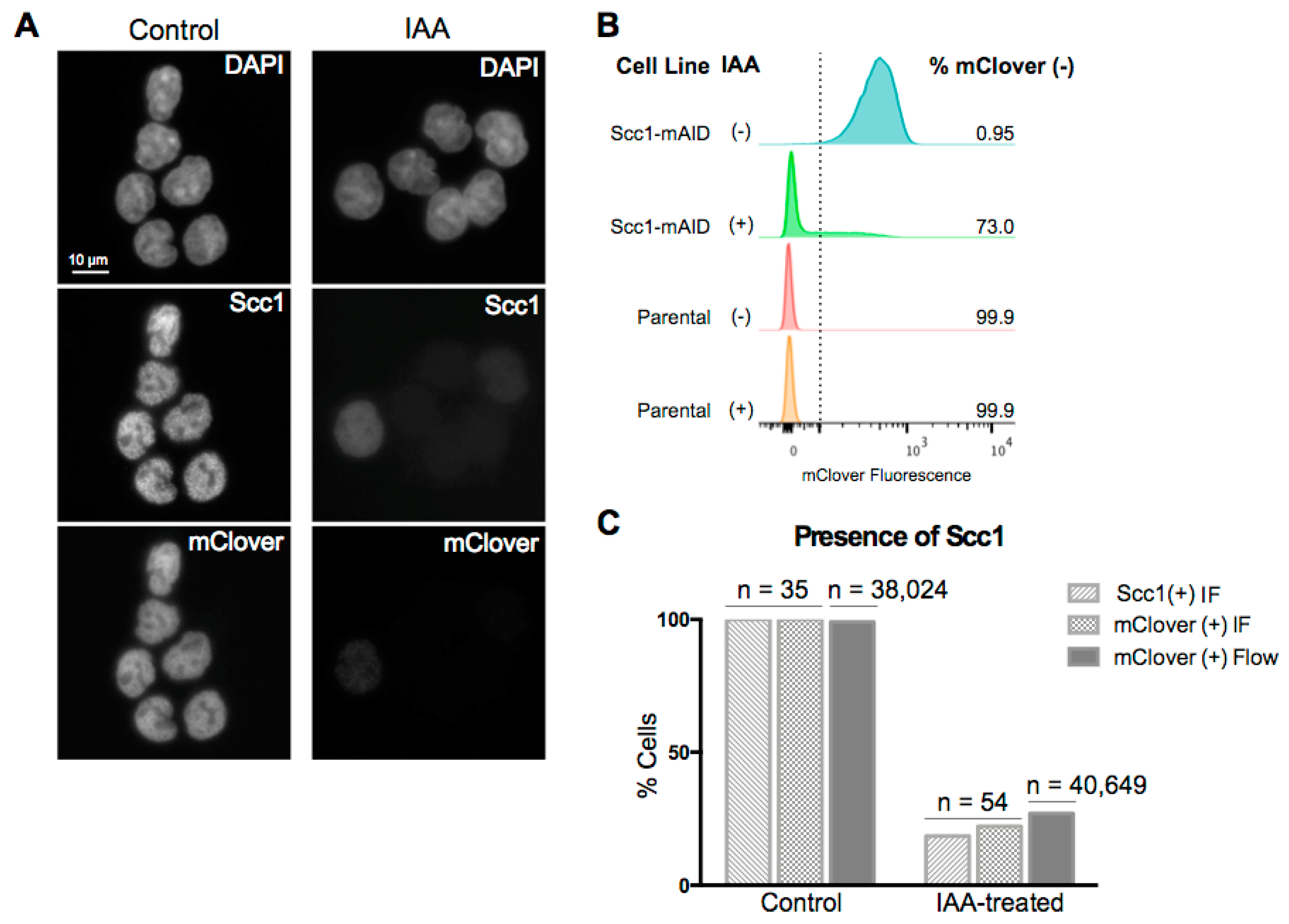
3.1. Efficient and Rapid Degradation of Endogenous Scc1
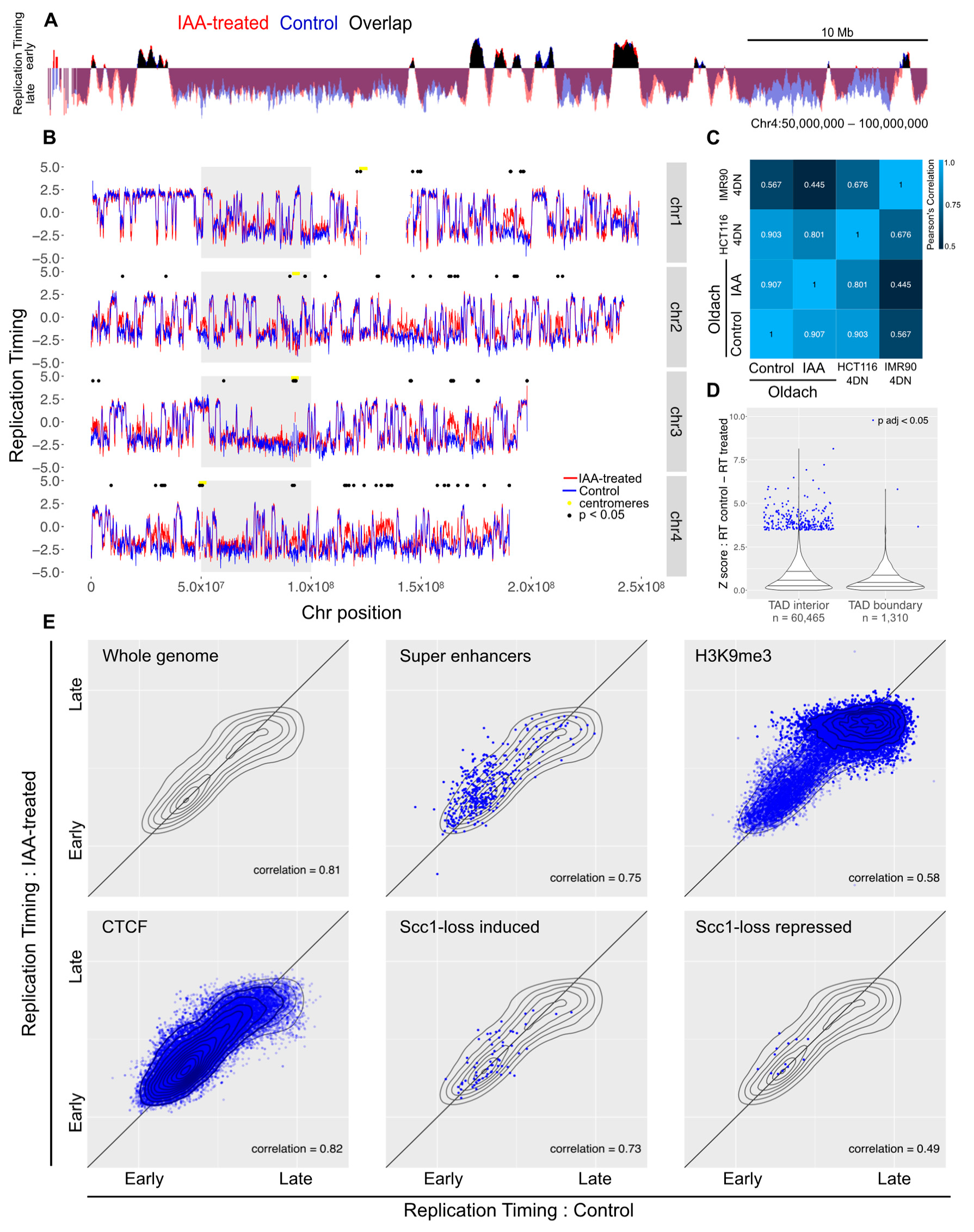
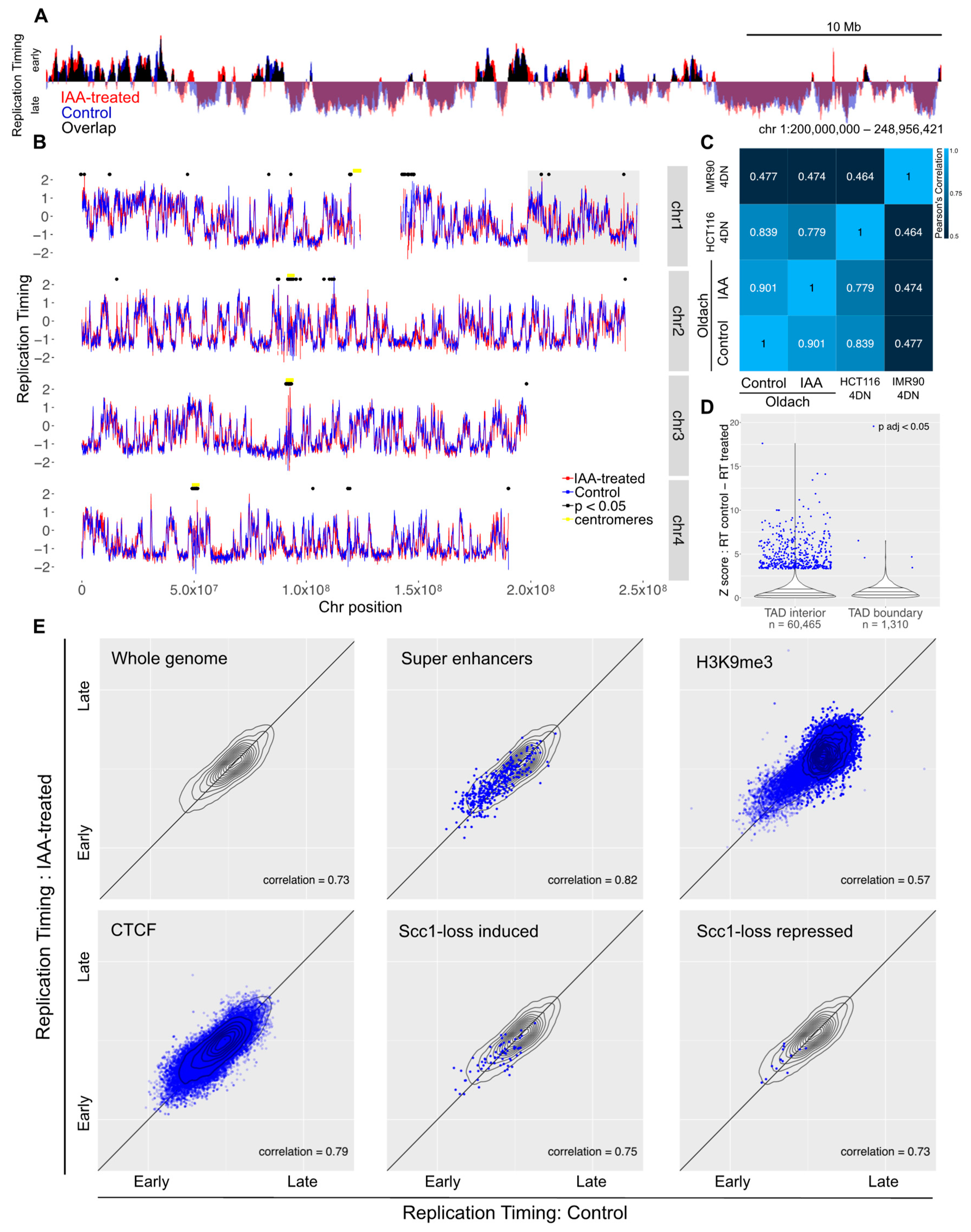
3.2. Acute Loss of Cohesin in S Phase Does Not Perturb Replication Patterns
3.3. Loss of Cohesin from Early G1 Does Not Perturb Replication Timing
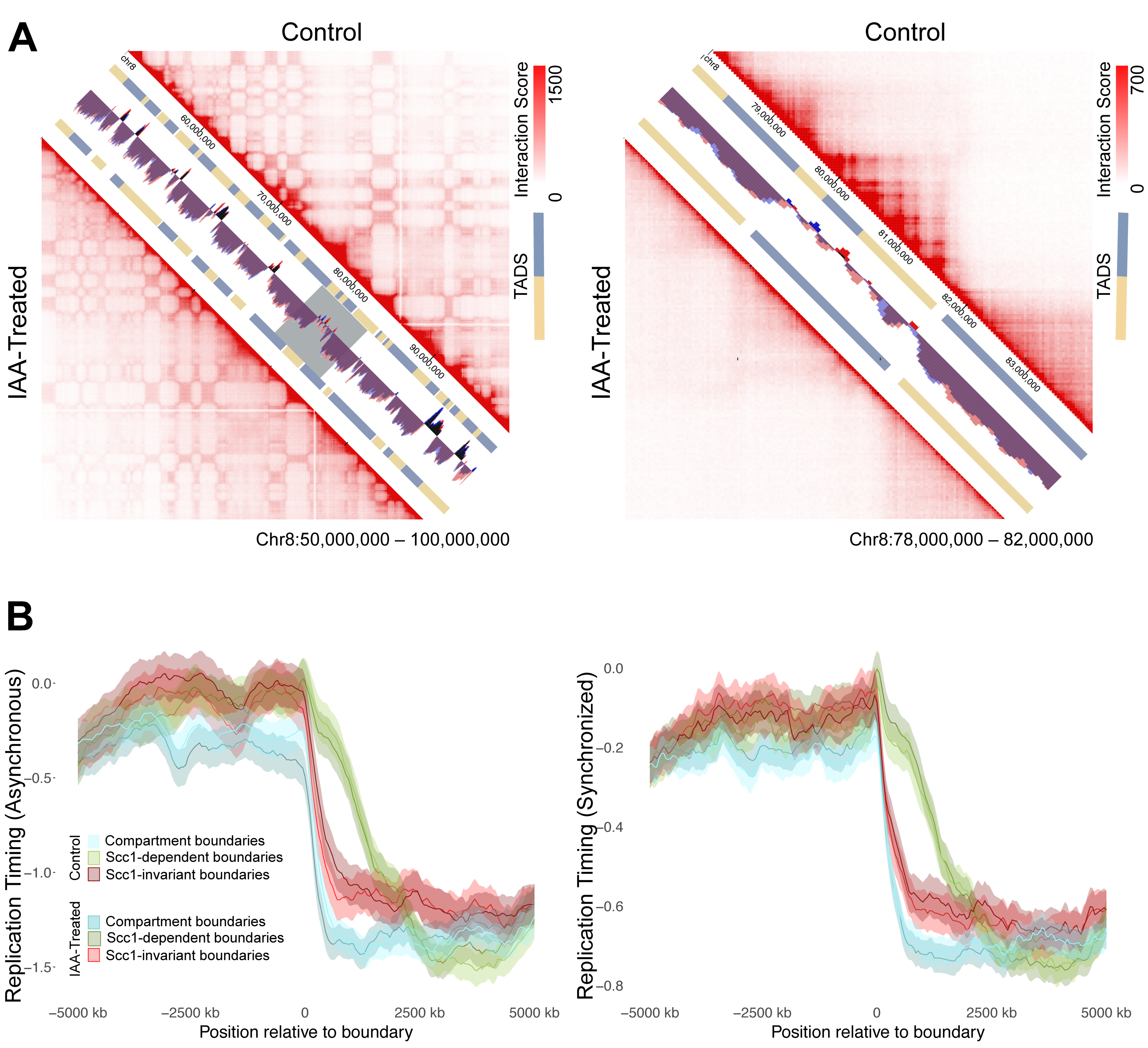
3.4. Impact of Genome Organization on Replication Timing
4. Discussion & Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Kahli, M.; D’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.-L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2015, 7, 10208. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Baris, A.; Aladjem, M.I. Replication timing and nuclear structure. Curr. Opin. Cell Biol. 2018, 52, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, D.; Denecker, T.; Maric, C.; Fauchereau, F.; Baldacci, G.; Cadoret, J.-C. Characterization of the replication timing program of 6 human model cell lines. Genomics Data 2016, 9, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Sasaki, T.; Battaglia, D.; Kulik, M.; Zhang, J.; Dalton, S.; Gilbert, D.M. Replication Timing: A Fingerprint for Cell Identity and Pluripotency. PLoS Comput. Biol. 2011, 7, e1002225. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Visser, A.E.; Eils, R.; Jauch, A.; Little, G.; Bakker, P.J.M.; Cremer, T.; Aten, J.A. Spatial Distributions of Early and Late Replicating Chromatin in Interphase Chromosome Territories. Exp. Cell Res. 1998, 243, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Berezney, R. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 2002, 115, 4037–4051. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.A.; Pombo, A. Replicon Clusters Are Stable Units of Chromosome Structure: Evidence that Nuclear Organization Contributes to the Efficient Activation and Propagation of S Phase in Human Cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Nakayasu, H.; Berezney, R. Mapping replicational sites in the eucaryotic cell nucleus. J. Cell Biol. 1989, 108, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Moindrot, B.; Audit, B.; Klous, P.; Baker, A.; Thermes, C.; De Laat, W.; Bouvet, P.; Mongelard, F.; Arneodo, A. 3D chromatin conformation correlates with replication timing and is conserved in resting cells. Nucleic Acids Res. 2012, 40, 9470–9481. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, E.; Farkash-Amar, S.; Polten, A.; Yakhini, Z.; Tanay, A.; Simon, I. Comparative Analysis of DNA Replication Timing Reveals Conserved Large-Scale Chromosomal Architecture. PLoS Genet. 2010, 6, e1001011. [Google Scholar] [CrossRef] [PubMed]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K. Disseminating the Genome: Joining, Resolving, and Separating Sister Chromatids During Mitosis and Meiosis. Annu. Rev. Genet. 2001, 35, 673–745. [Google Scholar] [CrossRef] [PubMed]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef] [PubMed]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Lubelsky, Y.; Prinz, J.A.; DeNapoli, L.; Li, Y.; Belsky, J.A.; MacAlpine, D.M. DNA replication and transcription programs respond to the same chromatin cues. Genome Res. 2014, 24, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Gilbert, D.M. The Spatial Position and Replication Timing of Chromosomal Domains are Both Established in Early G1 Phase. Mol. Cell 1999, 4, 983–993. [Google Scholar] [CrossRef]
- Dileep, V.; Ay, F.; Sima, J.; Vera, D.L.; Noble, W.S.; Gilbert, D.M. Topologically-associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication timing program. Genome Res. 2015, 25, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, J.M.; Danis, E.; Pasero, P.; Vassetzky, Y.; Méchali, M. Mitotic remodeling of the replicon and chromosome structure. Cell 2005, 123, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Dileep, V.; Rivera-Mulia, J.C.; Sima, J.; Gilbert, D.M. Large-Scale Chromatin Structure-Function Relationships during the Cell Cycle and Development: Insights from Replication Timing. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 2009, 6, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Natsume, T.; Kiyomitsu, T.; Saga, Y.; Kanemaki, M.T. Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 2016, 15, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.-C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.-R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305.e24–320.e24. [Google Scholar] [CrossRef] [PubMed]
- Marchal, C.; Sasaki, T.; Vera, D.; Wilson, K.; Sima, J.; Rivera-Mulia, J.C.; Trevilla-García, C.; Nogues, C.; Nafie, E.; Gilbert, D.M. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 2018, 13, 819–839. [Google Scholar] [CrossRef] [PubMed]
- Peace, J.M.; Villwock, S.K.; Zeytounian, J.L.; Gan, Y.; Aparicio, O.M. Quantitative BrdU immunoprecipitation method demonstrates that Fkh1 and Fkh2 are rate-limiting activators of replication origins that reprogram replication timing in G1 phase. Genome Res. 2016, 26, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-performance genomic feature operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef] [PubMed]
- Karimzadeh, M.; Ernst, C.; Kundaje, A.; Hoffman, M.M. Umap and Bismap: Quantifying genome and methylome mappability. Nucleic Acids Res. 2018, 46, e120. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, F.; Zhang, B.; Zhang, L.; Xu, J.; Kuang, D.; Li, D.; Choudhary, M.N.K.; Li, Y.; Hu, M.; et al. The 3D Genome Browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Weddington, N.; Stuy, A.; Hiratani, I.; Ryba, T.; Yokochi, T.; Gilbert, D.M. ReplicationDomain: A visualization tool and comparative database for genome-wide replication timing data. BMC Bioinforma. 2008, 9, 530. [Google Scholar] [CrossRef] [PubMed]
- Sima, J.; Chakraborty, A.; Dileep, V.; Michalski, M.; Rivera-Mulia, J.C.; Trevilla-Garcia, C.; Klein, K.N.; Bartlett, D.; Washburn, B.K.; Paulsen, M.T.; et al. Identification of cis elements for spatio-temporal control of DNA replication. Cell 2018, 176, 1–15. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Start | End | RT Control—RT IAA | |
|---|---|---|---|---|
| Asynchronous | Synchronous | |||
| chr3 | 198100000 | 198150000 | −3.440337 | 3.242949 |
| chr4 | 49300000 | 49350000 | 2.723062 | 2.296154 |
| chr4 | 119400000 | 119450000 | 3.486406 | −1.410933 |
| chr4 | 190000000 | 190050000 | 4.479181 | −1.897292 |
| chr4 | 190050000 | 190100000 | 12.678844 | −5.136778 |
| chr4 | 190100000 | 190150000 | 25.976415 | −10.545368 |
| chr5 | 49650000 | 49700000 | −3.258203 | 4.272367 |
| chr5 | 181250000 | 181300000 | 2.921178 | −1.623559 |
| chr7 | 56850000 | 56900000 | 2.779265 | 1.355865 |
| chr9 | 150000 | 200000 | 3.343623 | −1.447844 |
| chr13 | 114300000 | 114350000 | 3.460791 | 1.561016 |
| chr13 | 114350000 | 114364328 | 7.856382 | 3.064328 |
| chr16 | 90100000 | 90150000 | 4.566427 | −3.221925 |
| chr21 | 6350000 | 6400000 | 3.521525 | −2.910266 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oldach, P.; Nieduszynski, C.A. Cohesin-Mediated Genome Architecture Does Not Define DNA Replication Timing Domains. Genes 2019, 10, 196. https://doi.org/10.3390/genes10030196
Oldach P, Nieduszynski CA. Cohesin-Mediated Genome Architecture Does Not Define DNA Replication Timing Domains. Genes. 2019; 10(3):196. https://doi.org/10.3390/genes10030196
Chicago/Turabian StyleOldach, Phoebe, and Conrad A. Nieduszynski. 2019. "Cohesin-Mediated Genome Architecture Does Not Define DNA Replication Timing Domains" Genes 10, no. 3: 196. https://doi.org/10.3390/genes10030196
APA StyleOldach, P., & Nieduszynski, C. A. (2019). Cohesin-Mediated Genome Architecture Does Not Define DNA Replication Timing Domains. Genes, 10(3), 196. https://doi.org/10.3390/genes10030196