Origin Firing Regulations to Control Genome Replication Timing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. A Simplified Model of Genome Replication in Higher Eukaryotes
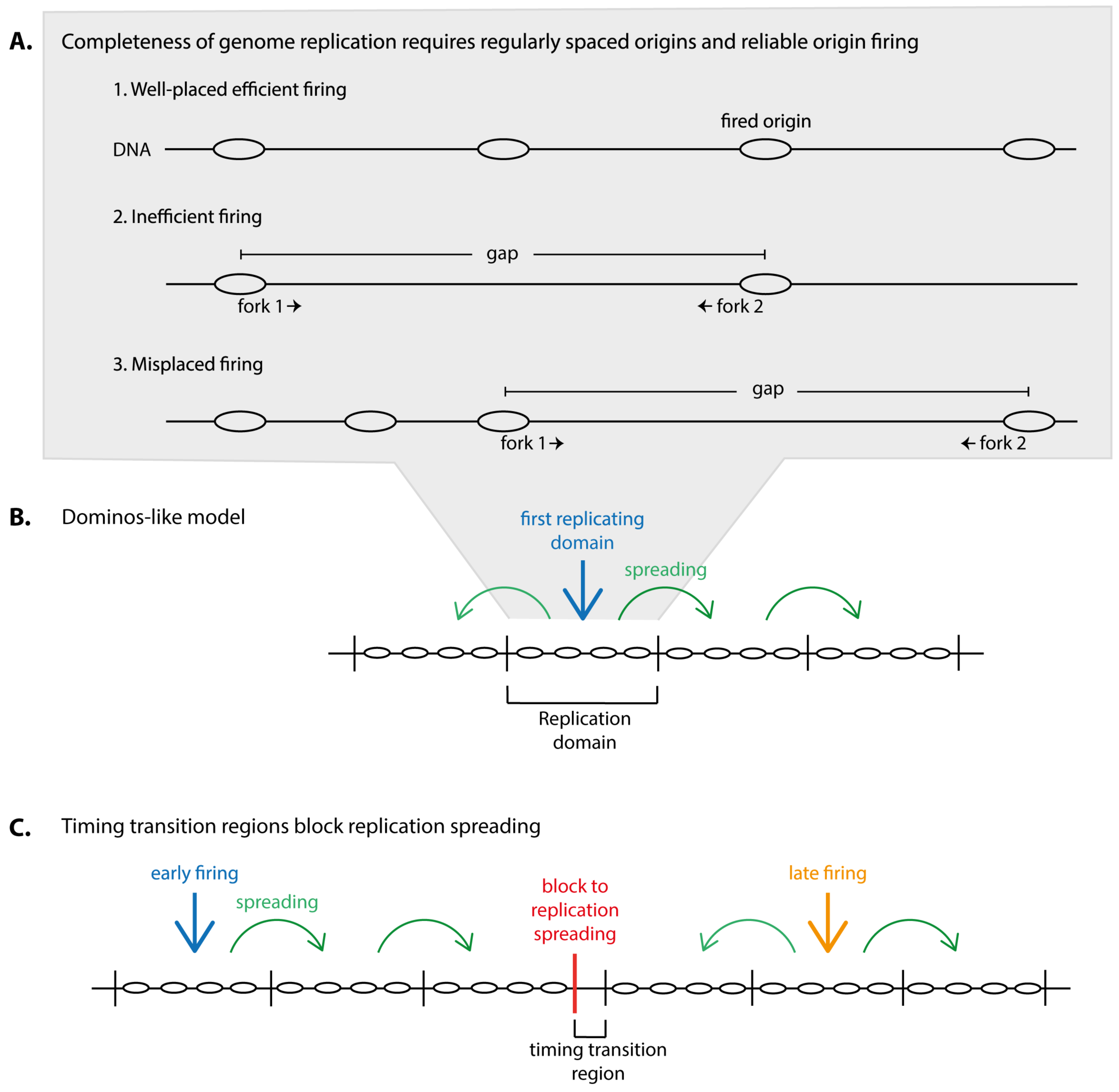
1.1. Completeness of Genome Replication Requires Small Distances between Origins and Reliable Origin Firing
1.2. Domain-Wise Replication May Help Complete Genome Duplication in Limited Origin Firing Conditions
1.3. Timing of Domain Replication
2. Complete and Well-Timed Domain-Wise Genome Replication Requires Coherent Origin Firing within Domains and Proper Control of Origin Firing Time
3. Molecular Mechanisms of Origin Firing
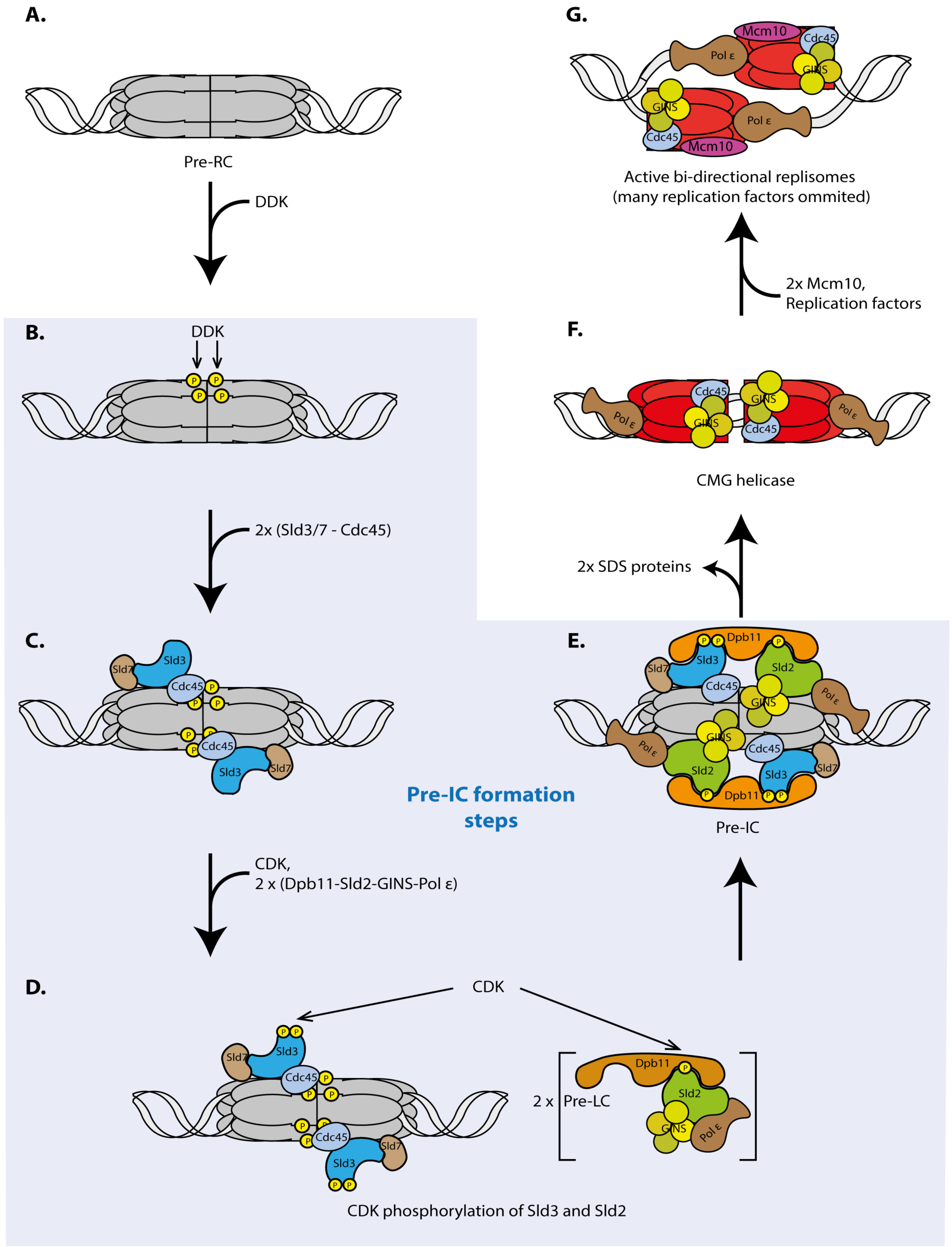
4. Pre-IC Formation is a Main Regulation Step of Origin Firing
5. Origin Firing Control to Determine Replication Timing
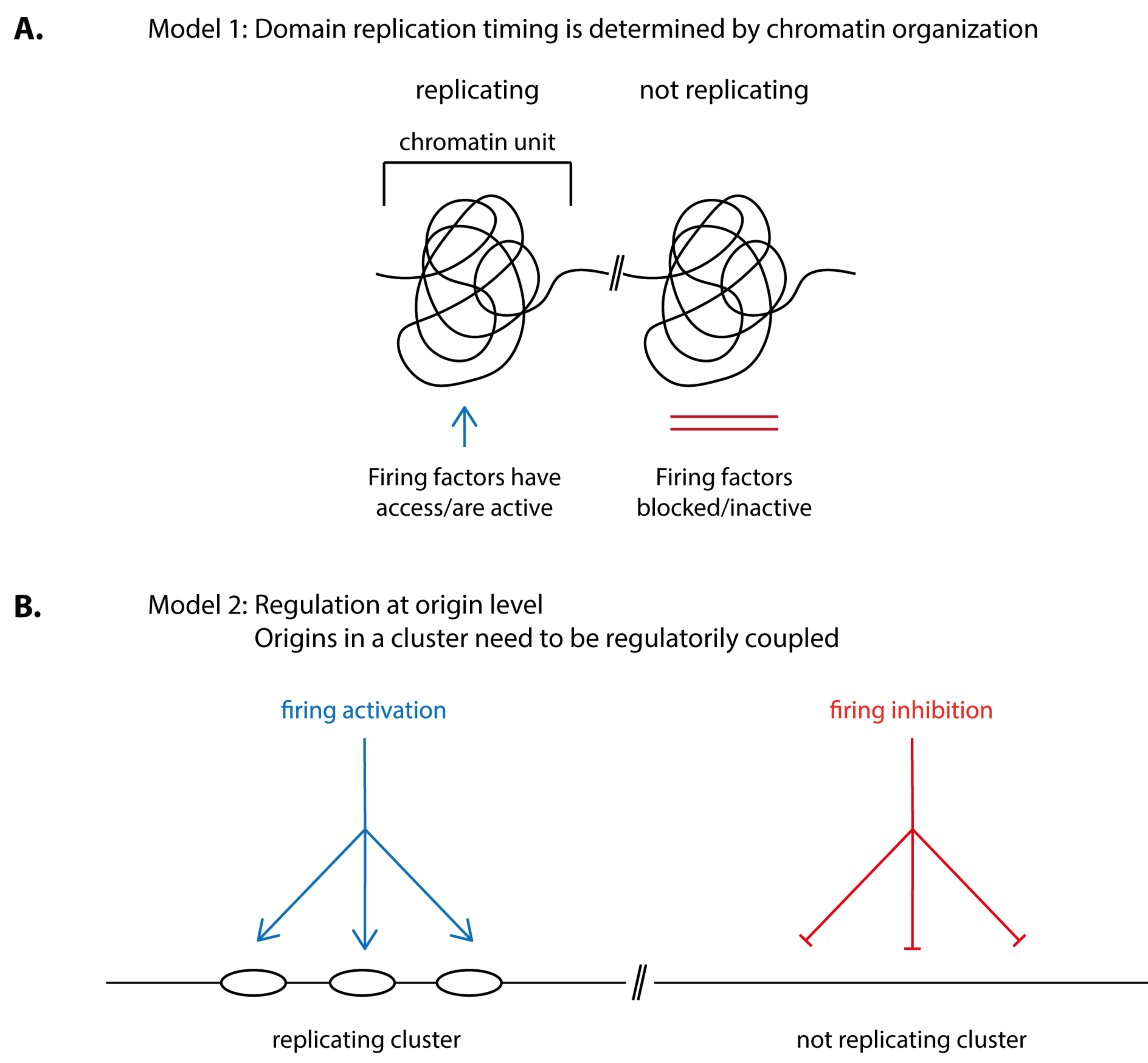
5.1. Structural Chromatin Units May Underlie Replication Timing
5.2. How Could the Folding of Chromatin into Physical Units Determine Origin Firing Time?
5.3. In Yeast, Several Origin Firing Control Pathways in Combination with Competition between Origins for Firing Factors Determine Replication Time
5.4. Forkhead Transcription Factors Advance Origin Firing Timing in Budding Yeast
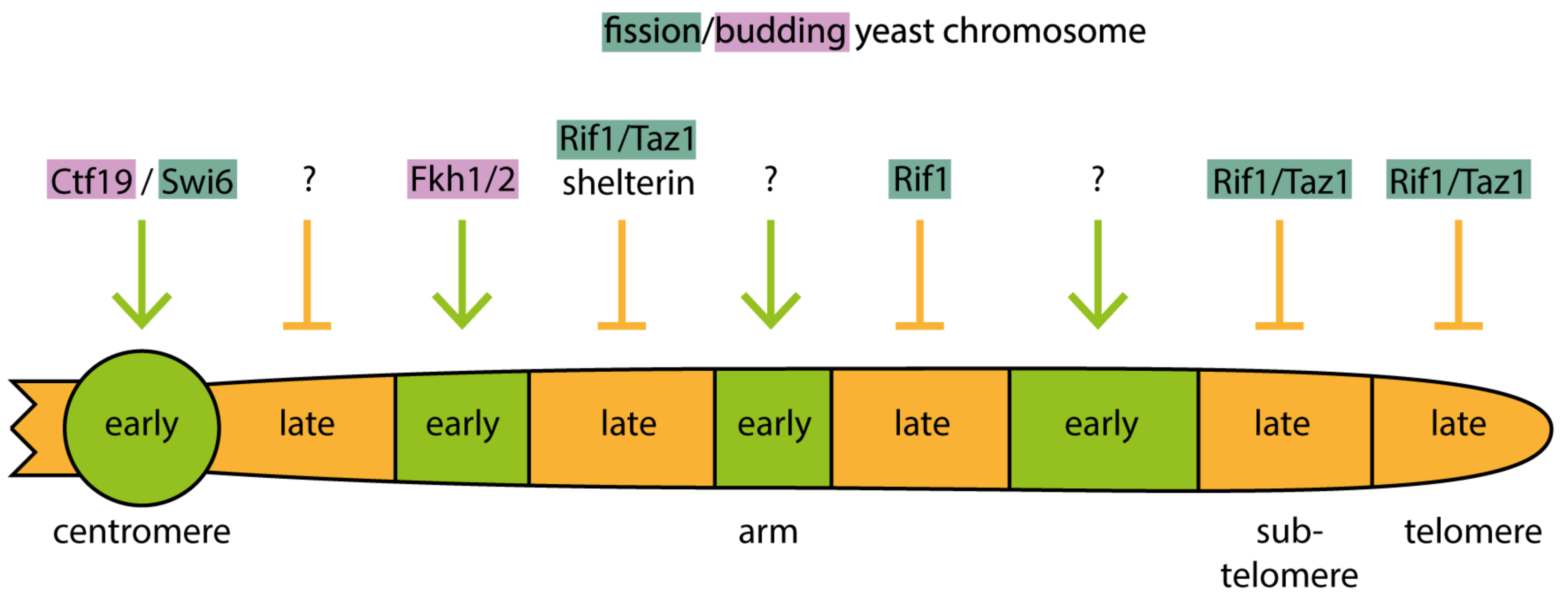
5.5. Early Firing of Pericentromeric Heterochromatin Origins Depends on the Recruitment of DDK by the Ctf19 Kinetochore Protein or Swi6 in Budding and Fission Yeast, Respectively
5.6. Rif1 and Taz1 Facilitate Late Origin Firing in Yeasts
5.7. Metazoa May Utilise Rif1-Dependent and Independent Pathways to Mediate Late Replication
5.8. The TMT Complex May Help Control Firing Timing in Metazoa
6. Firing Coherence Mechanisms
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leonard, A.C.; Mechali, M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013, 5, a010116. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef]
- Burkhart, R.; Schulte, D.; Hu, D.; Musahl, C.; Gohring, F.; Knippers, R. Interactions of human nuclear proteins P1Mcm3 and P1Cdc46. Eur. J. Biochem. 1995, 228, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Kawasaki, Y.; Tye, B.K. Physical interactions among MCM proteins and effects of MCM dosage on DNA-replication in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 5081–5090. [Google Scholar] [CrossRef] [PubMed]
- Rowles, A.; Chong, J.P.; Brown, L.; Howell, M.; Evan, G.I.; Blow, J.J. Interaction between the origin recognition complex and the replication licensing system in Xenopus. Cell 1996, 87, 287–296. [Google Scholar] [CrossRef]
- Donovan, S.; Harwood, J.; Drury, L.S.; Diffley, J.F.X. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 5611–5616. [Google Scholar] [CrossRef] [PubMed]
- Mahbubani, H.M.; Chong, J.P.; Chevalier, S.; Thommes, P.; Blow, J.J. Cell cycle regulation of the replication licensing system: Involvement of a Cdk-dependent inhibitor. J. Cell Biol. 1997, 136, 125–135. [Google Scholar] [CrossRef]
- Edwards, M.C.; Tutter, A.V.; Cvetic, C.; Gilbert, C.H.; Prokhorova, T.A.; Walter, J.C. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 2002, 277, 33049–33057. [Google Scholar] [CrossRef]
- Santocanale, C.; Sharma, K.; Diffley, J.F.X. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999, 13, 2360–2364. [Google Scholar] [CrossRef]
- Woodward, A.M.; Gohler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Mackenzie, A.; Donaldson, A.; Zegerman, P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Collart, C.; Allen, G.E.; Bradshaw, C.R.; Smith, J.C.; Zegerman, P. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 2013, 341, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.G.; Winter, S.L.; Zaika, E.; Cao, T.V.; Oguz, U.; Koomen, J.M.; Hamlin, J.L.; Alexandrow, M.G. Cdc45 limits replicon usage from a low density of preRCs in mammalian cells. PLoS ONE 2011, 6, e17533. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F.X. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.W.; Lyou, Y.; Townes, T.M.; Schubeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachages, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Sporbert, A.; Gahl, A.; Ankerhold, R.; Leonhardt, H.; Cardoso, M.C. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol. Cell 2002, 10, 1355–1365. [Google Scholar] [CrossRef]
- Sadoni, N.; Cardoso, M.C.; Stelzer, E.H.; Leonhardt, H.; Zink, D. Stable chromosomal units determine the spatial and temporal organization of DNA replication. J. Cell Sci. 2004, 117, 5353–5365. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Olivares-Chauvet, P.; Shaw, A.; Jackson, D.A. S phase progression in human cells is dictated by the genetic continuity of DNA foci. PLoS Genet. 2010, 6, e1000900. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, G.; Rappailles, A.; Baker, A.; Chen, C.L.; Arneodo, A.; Goldar, A.; d’Aubenton-Carafa, Y.; Thermes, C.; Audit, B.; Hyrien, O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011, 7, e1002322. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Morita, T.; Sato, C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res. 1986, 165, 291–297. [Google Scholar] [CrossRef]
- Dimitrova, D.S.; Gilbert, D.M. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell 1999, 4, 983–993. [Google Scholar] [CrossRef]
- Ma, H.; Samarabandu, J.; Devdhar, R.S.; Acharya, R.; Cheng, P.C.; Meng, C.; Berezney, R. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol. 1998, 143, 1415–1425. [Google Scholar] [CrossRef]
- Leonhardt, H.; Rahn, H.P.; Weinzierl, P.; Sporbert, A.; Cremer, T.; Zink, D.; Cardoso, M.C. Dynamics of DNA replication factories in living cells. J. Cell Biol. 2000, 149, 271–280. [Google Scholar] [CrossRef]
- Wansink, D.G.; Manders, E.E.; van der Kraan, I.; Aten, J.A.; van Driel, R.; de Jong, L. RNA polymerase II transcription is concentrated outside replication domains throughout S-phase. J. Cell Sci. 1994, 107 Pt 6, 1449–1456. [Google Scholar]
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Meryet-Figuiere, M.; Alaei-Mahabadi, B.; Ali, M.M.; Mitra, S.; Subhash, S.; Pandey, G.K.; Larsson, E.; Kanduri, C. Temporal separation of replication and transcription during S-phase progression. Cell Cycle 2014, 13, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; De, S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res. 2013, 41, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Lawrence, M.S.; Carter, S.L.; Stewart, C.; Gabriel, S.B.; Lander, E.S.; Meyerson, M.; Beroukhim, R.; Getz, G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013, 23, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Natsume, T.; Muller, C.A.; Katou, Y.; Retkute, R.; Gierlinski, M.; Araki, H.; Blow, J.J.; Shirahige, K.; Nieduszynski, C.A.; Tanaka, T.U. Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Mol. Cell 2013, 50, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Takahashi, T.S.; Nakagawa, T.; Nakayama, J.; Masukata, H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat. Cell Biol. 2009, 11, 357–362. [Google Scholar] [CrossRef]
- Jackson, D.A.; Pombo, A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 2015, 519, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Diffley, J.F.X. Regulation of early events in chromosome replication. Curr. Biol. 2004, 14, R778–786. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F.X. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Matsui, T.; Takisawa, H.; Smith, M.M. Regulation of replication licensing by acetyltransferase Hbo1. Mol. Cell Biol. 2006, 26, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Miotto, B.; Struhl, K. HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev. 2008, 22, 2633–2638. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Yugawa, T.; Iizuka, M.; Kiyono, T.; Fujita, M. Chromatin remodeler sucrose nonfermenting 2 homolog (SNF2H) is recruited onto DNA replication origins through interaction with Cdc10 protein-dependent transcript 1 (Cdt1) and promotes pre-replication complex formation. J. Biol. Chem. 2011, 286, 39200–39210. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Yasukouchi, S.; Osano, S.; Watanabe, S.; Aizawa, M.; Yugawa, T.; Kiyono, T.; Kurumizaka, H.; et al. Cdt1-binding protein GRWD1 is a novel histone-binding protein that facilitates MCM loading through its influence on chromatin architecture. Nucleic Acids Res. 2015, 43, 5898–5911. [Google Scholar] [CrossRef] [PubMed]
- Tardat, M.; Murr, R.; Herceg, Z.; Sardet, C.; Julien, E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 2007, 179, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.; Elvers, I.; Trelle, M.B.; Menzel, T.; Eskildsen, M.; Jensen, O.N.; Helleday, T.; Helin, K.; Sorensen, C.S. The histone methyltransferase SET8 is required for S-phase progression. J. Cell Biol. 2007, 179, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Sy, S.M.; van Deursen, J.M.; Chen, J. Direct interaction between SET8 and proliferating cell nuclear antigen couples H4-K20 methylation with DNA replication. J. Biol. Chem. 2008, 283, 11073–11077. [Google Scholar] [CrossRef] [PubMed]
- Centore, R.C.; Havens, C.G.; Manning, A.L.; Li, J.M.; Flynn, R.L.; Tse, A.; Jin, J.; Dyson, N.J.; Walter, J.C.; Zou, L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 2010, 40, 22–33. [Google Scholar] [CrossRef]
- Tardat, M.; Brustel, J.; Kirsh, O.; Lefevbre, C.; Callanan, M.; Sardet, C.; Julien, E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 2010, 12, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Frappier, L.; Mechali, M. MCM-BP regulates unloading of the MCM2-7 helicase in late S phase. Genes Dev. 2011, 25, 165–175. [Google Scholar] [CrossRef]
- Ding, L.; Forsburg, S.L. Schizosaccharomyces pombe minichromosome maintenance-binding protein (MCM-BP) antagonizes MCM helicase. J. Biol. Chem. 2011, 286, 32918–32930. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.E.; Lewis, P.W.; Botchan, M.R. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 2006, 103, 10236–10241. [Google Scholar] [CrossRef] [PubMed]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Abid Ali, F.; Douglas, M.E.; Locke, J.; Pye, V.E.; Nans, A.; Diffley, J.F.X.; Costa, A. Cryo-EM structure of a licensed DNA replication origin. Nat. Commun. 2017, 8, 2241. [Google Scholar] [CrossRef]
- Sheu, Y.J.; Stillman, B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 2006, 24, 101–113. [Google Scholar] [CrossRef]
- Francis, L.I.; Randell, J.C.; Takara, T.J.; Uchima, L.; Bell, S.P. Incorporation into the prereplicative complex activates the Mcm2-7 helicase for Cdc7-Dbf4 phosphorylation. Genes Dev. 2009, 23, 643–654. [Google Scholar] [CrossRef]
- Sheu, Y.J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Yeeles, J.T.; Diffley, J.F. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016, 35, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Tak, Y.S.; Sugino, A.; Araki, H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J. 2001, 20, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa-Onami, M.; Araki, H.; Tanaka, S. Pre-initiation complex assembly functions as a molecular switch that splits the Mcm2-7 double hexamer. EMBO Rep. 2017, 18, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, Y.; Masumoto, H.; Sugino, A.; Araki, H. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 1998, 18, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, H.; Muramatsu, S.; Kamimura, Y.; Araki, H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature 2002, 415, 651–655. [Google Scholar] [CrossRef]
- Zegerman, P.; Diffley, J.F.X. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 2007, 445, 281–285. [Google Scholar] [CrossRef]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef]
- Muramatsu, S.; Hirai, K.; Tak, Y.S.; Kamimura, Y.; Araki, H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes Dev. 2010, 24, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Stillman, B. Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science 1998, 280, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Kanemaki, M.; Labib, K. Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO J. 2006, 25, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Jones, R.C.; Sanchez-Diaz, A.; Kanemaki, M.; van Deursen, F.; Edmondson, R.D.; Labib, K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006, 8, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Labib, K.; Tercero, J.A.; Diffley, J.F.X. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 2000, 288, 1643–1647. [Google Scholar] [CrossRef]
- Kanke, M.; Kodama, Y.; Takahashi, T.S.; Nakagawa, T.; Masukata, H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 2012, 31, 2182–2194. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, F.; Sengupta, S.; De Piccoli, G.; Sanchez-Diaz, A.; Labib, K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 2012, 31, 2195–2206. [Google Scholar] [CrossRef]
- Watase, G.; Takisawa, H.; Kanemaki, M.T. Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Curr. Biol. 2012, 22, 343–349. [Google Scholar] [CrossRef]
- Boos, D.; Frigola, J.; Diffley, J.F.X. Activation of the replicative DNA helicase: Breaking up is hard to do. Curr. Opin. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Sansam, C.L.; Cruz, N.M.; Danielian, P.S.; Amsterdam, A.; Lau, M.L.; Hopkins, N.; Lees, J.A. A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev. 2010, 24, 183–194. [Google Scholar] [CrossRef]
- Kumagai, A.; Shevchenko, A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pulido, L.; Diffley, J.F.X.; Ponting, C.P. Homology explains the functional similarities of Treslin/Ticrr and Sld3. Curr. Biol. 2010, 20, R509–R510. [Google Scholar] [CrossRef] [PubMed]
- Boos, D.; Sanchez-Pulido, L.; Rappas, M.; Pearl, L.H.; Oliver, A.W.; Ponting, C.P.; Diffley, J.F.X. Regulation of DNA Replication through Sld3-Dpb11 Interaction Is Conserved from Yeast to Humans. Curr. Biol. 2011, 21, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol. Cell Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef]
- Tanaka, T.; Umemori, T.; Endo, S.; Muramatsu, S.; Kanemaki, M.; Kamimura, Y.; Obuse, C.; Araki, H. Sld7, an Sld3-associated protein required for efficient chromosomal DNA replication in budding yeast. EMBO J. 2011. [Google Scholar] [CrossRef]
- Boos, D.; Yekezare, M.; Diffley, J.F. Identification of a heteromeric complex that promotes DNA replication origin firing in human cells. Science 2013, 340, 981–984. [Google Scholar] [CrossRef]
- Kohler, K.; Sanchez-Pulido, L.; Hofer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-cyclin C transcription regulator functions in genome replication through metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef]
- Walter, J.C. Evidence for sequential action of cdc7 and cdk2 protein kinases during initiation of DNA replication in Xenopus egg extracts. J. Biol. Chem. 2000, 275, 39773–39778. [Google Scholar] [CrossRef]
- Zegerman, P.; Diffley, J.F.X. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mosqueda, J.; Maas, N.L.; Jonsson, Z.O.; Defazio-Eli, L.G.; Wohlschlegel, J.; Toczyski, D.P. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 2010, 467, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Duch, A.; Palou, G.; Jonsson, Z.O.; Palou, R.; Calvo, E.; Wohlschlegel, J.; Quintana, D.G. A Dbf4 mutant contributes to bypassing the Rad53-mediated block of origins of replication in response to genotoxic stress. J. Biol. Chem. 2011, 286, 2486–2491. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Kumagai, A.; Schlacher, K.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol. Cell 2015, 57, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Sansam, C.G.; Pietrzak, K.; Majchrzycka, B.; Kerlin, M.A.; Chen, J.; Rankin, S.; Sansam, C.L. A mechanism for epigenetic control of DNA replication. Genes Dev. 2018, 32, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N. DNA replication timing: random thoughts about origin firing. Nat. Cell Biol. 2006, 8, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Sparvoli, E.; Levi, M.; Rossi, E. Replicon clusters may form structurally stable complexes of chromatin and chromosomes. J. Cell Sci. 1994, 107 Pt 11, 3097–3103. [Google Scholar]
- Zink, D.; Cremer, T.; Saffrich, R.; Fischer, R.; Trendelenburg, M.F.; Ansorge, W.; Stelzer, E.H. Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 1998, 102, 241–251. [Google Scholar] [CrossRef]
- Yaffe, E.; Farkash-Amar, S.; Polten, A.; Yakhini, Z.; Tanay, A.; Simon, I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010, 6, e1001011. [Google Scholar] [CrossRef]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef]
- Dileep, V.; Ay, F.; Sima, J.; Vera, D.L.; Noble, W.S.; Gilbert, D.M. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 2015, 25, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, F.; Murphy, C.S.; Davidson, M.W.; Gilbert, D.M. G2 phase chromatin lacks determinants of replication timing. J. Cell Biol. 2010, 189, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Knott, S.R.; Viggiani, C.J.; Tavare, S.; Aparicio, O.M. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev. 2009, 23, 1077–1090. [Google Scholar] [CrossRef]
- Aparicio, J.G.; Viggiani, C.J.; Gibson, D.G.; Aparicio, O.M. The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol. Cell Biol. 2004, 24, 4769–4780. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Bacal, J.; Desmarais, D.; Padioleau, I.; Tsaponina, O.; Chabes, A.; Pantesco, V.; Dubois, E.; Parrinello, H.; Skrzypczak, M.; et al. The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol. Cell 2014, 54, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Tabib, A.; Hecht, M.; Cedar, H. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev. 2008, 22, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Koryakov, D.E.; Pokholkova, G.V.; Maksimov, D.A.; Belyakin, S.N.; Belyaeva, E.S.; Zhimulev, I.F. Induced transcription results in local changes in chromatin structure, replication timing, and DNA polytenization in a site of intercalary heterochromatin. Chromosoma 2012, 121, 573–583. [Google Scholar] [CrossRef]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef]
- Cosgrove, A.J.; Nieduszynski, C.A.; Donaldson, A.D. Ku complex controls the replication time of DNA in telomere regions. Genes Dev. 2002, 16, 2485–2490. [Google Scholar] [CrossRef]
- Ebrahimi, H.; Robertson, E.D.; Taddei, A.; Gasser, S.M.; Donaldson, A.D.; Hiraga, S. Early initiation of a replication origin tethered at the nuclear periphery. J. Cell Sci. 2010, 123, 1015–1019. [Google Scholar] [CrossRef]
- Knott, S.R.; Peace, J.M.; Ostrow, A.Z.; Gan, Y.; Rex, A.E.; Viggiani, C.J.; Tavare, S.; Aparicio, O.M. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell 2012, 148, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Tazumi, A.; Fukuura, M.; Nakato, R.; Kishimoto, A.; Takenaka, T.; Ogawa, S.; Song, J.H.; Takahashi, T.S.; Nakagawa, T.; Shirahige, K.; et al. Telomere-binding protein Taz1 controls global replication timing through its localization near late replication origins in fission yeast. Genes Dev. 2012, 26, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Kido, S.; Handa, T.; Ogawa, H.; Asakawa, H.; Takahashi, T.S.; Nakagawa, T.; Hiraoka, Y.; Masukata, H. Shelterin promotes tethering of late replication origins to telomeres for replication-timing control. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Lengronne, A.; Shi, D.; Forey, R.; Skrzypczak, M.; Ginalski, K.; Yan, C.; Wang, X.; Cao, Q.; Pasero, P.; et al. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. 2017, 31, 2405–2415. [Google Scholar] [CrossRef]
- Hiraga, S.; Alvino, G.M.; Chang, F.; Lian, H.Y.; Sridhar, A.; Kubota, T.; Brewer, B.J.; Weinreich, M.; Raghuraman, M.K.; Donaldson, A.D. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 2014, 28, 372–383. [Google Scholar] [CrossRef]
- Dave, A.; Cooley, C.; Garg, M.; Bianchi, A. Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep. 2014, 7, 53–61. [Google Scholar] [CrossRef]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thoma, N.H.; Hardy, C.F.; Shore, D. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef]
- Chikashige, Y.; Yamane, M.; Okamasa, K.; Tsutsumi, C.; Kojidani, T.; Sato, M.; Haraguchi, T.; Hiraoka, Y. Membrane proteins Bqt3 and -4 anchor telomeres to the nuclear envelope to ensure chromosomal bouquet formation. J. Cell Biol. 2009, 187, 413–427. [Google Scholar] [CrossRef]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Sukackaite, R.; Cornacchia, D.; Jensen, M.R.; Mas, P.J.; Blackledge, M.; Enervald, E.; Duan, G.; Auchynnikava, T.; Kohn, M.; Hart, D.J.; et al. Mouse Rif1 is a regulatory subunit of protein phosphatase 1 (PP1). Sci. Rep. 2017, 7, 2119. [Google Scholar] [CrossRef] [PubMed]
- Alver, R.C.; Chadha, G.S.; Gillespie, P.J.; Blow, J.J. Reversal of DDK-Mediated MCM Phosphorylation by Rif1-PP1 Regulates Replication Initiation and Replisome Stability Independently of ATR/Chk1. Cell Rep. 2017, 18, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.S. A role for DNA polymerase theta in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Araki, H. Multiple regulatory mechanisms to inhibit untimely initiation of DNA replication are important for stable genome maintenance. PLoS Genet. 2011, 7, e1002136. [Google Scholar] [CrossRef] [PubMed]
- Reusswig, K.U.; Zimmermann, F.; Galanti, L.; Pfander, B. Robust Replication Control Is Generated by Temporal Gaps between Licensing and Firing Phases and Depends on Degradation of Firing Factor Sld2. Cell Rep. 2016, 17, 556–569. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.H.; Leng, W.; Jung, S.Y.; Zhang, H.; Deng, M.; Evans, D.; Li, Y.; Luo, K.; Qin, B.; et al. A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Mol. Cell 2014, 56, 681–695. [Google Scholar] [CrossRef]
- Thomson, A.M.; Gillespie, P.J.; Blow, J.J. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J. Cell Biol. 2010, 188, 209–221. [Google Scholar] [CrossRef]
- Brummer, A.; Salazar, C.; Zinzalla, V.; Alberghina, L.; Hofer, T. Mathematical modelling of DNA replication reveals a trade-off between coherence of origin activation and robustness against rereplication. PLoS Comput. Biol. 2010, 6, e1000783. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Ferrell, J.E., Jr. Multisite phosphorylation and the countdown to S phase. Cell 2001, 107, 819–822. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boos, D.; Ferreira, P. Origin Firing Regulations to Control Genome Replication Timing. Genes 2019, 10, 199. https://doi.org/10.3390/genes10030199
Boos D, Ferreira P. Origin Firing Regulations to Control Genome Replication Timing. Genes. 2019; 10(3):199. https://doi.org/10.3390/genes10030199
Chicago/Turabian StyleBoos, Dominik, and Pedro Ferreira. 2019. "Origin Firing Regulations to Control Genome Replication Timing" Genes 10, no. 3: 199. https://doi.org/10.3390/genes10030199
APA StyleBoos, D., & Ferreira, P. (2019). Origin Firing Regulations to Control Genome Replication Timing. Genes, 10(3), 199. https://doi.org/10.3390/genes10030199