Classifying Breast Cancer Subtypes Using Multiple Kernel Learning Based on Omics Data

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Data Sources
2.2. Feature Selection
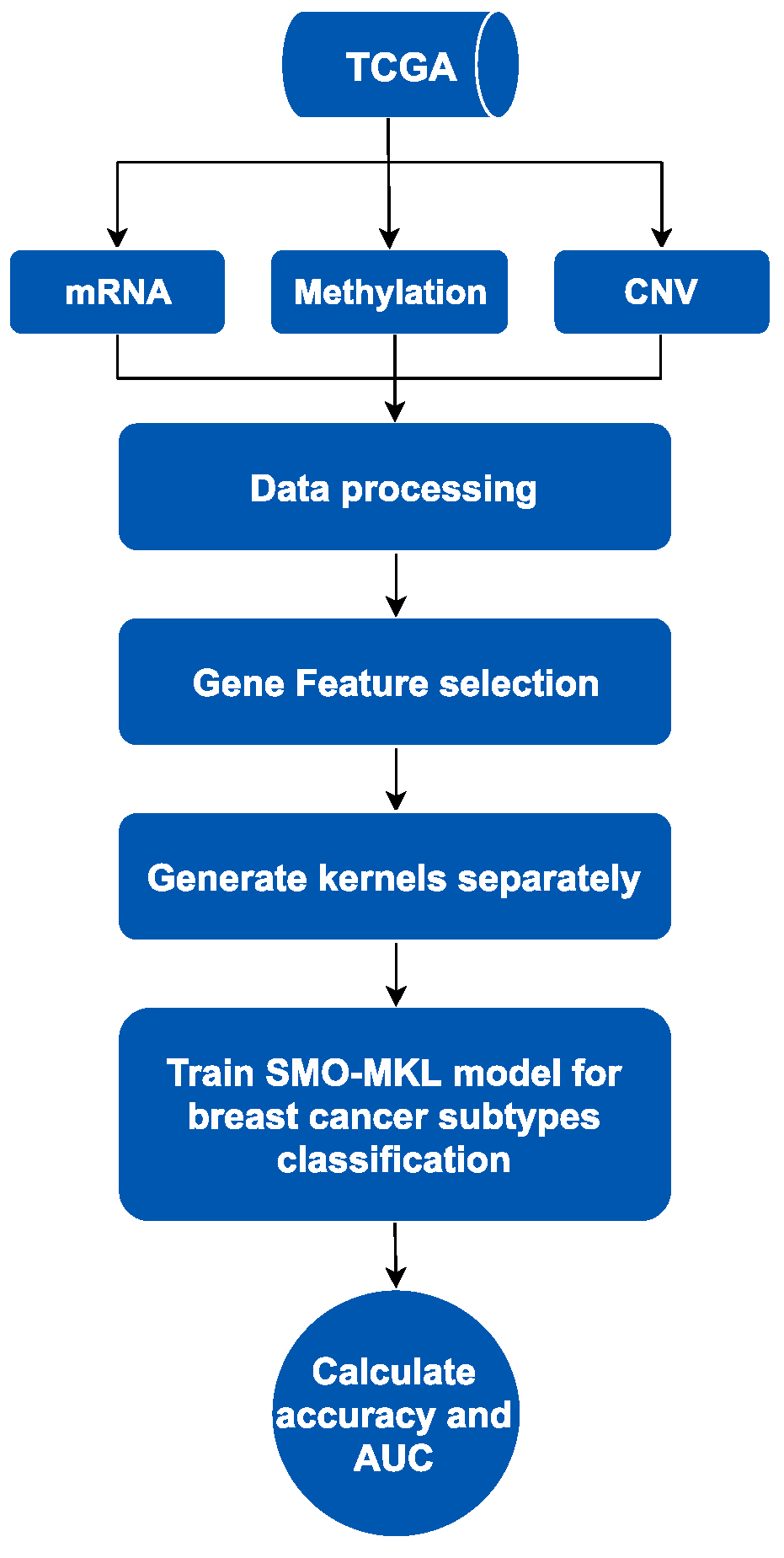
2.3. The Process of Classification
2.3.1. Multi-Omics Data Fusing
2.3.2. SMO-MKL Classification
3. Results
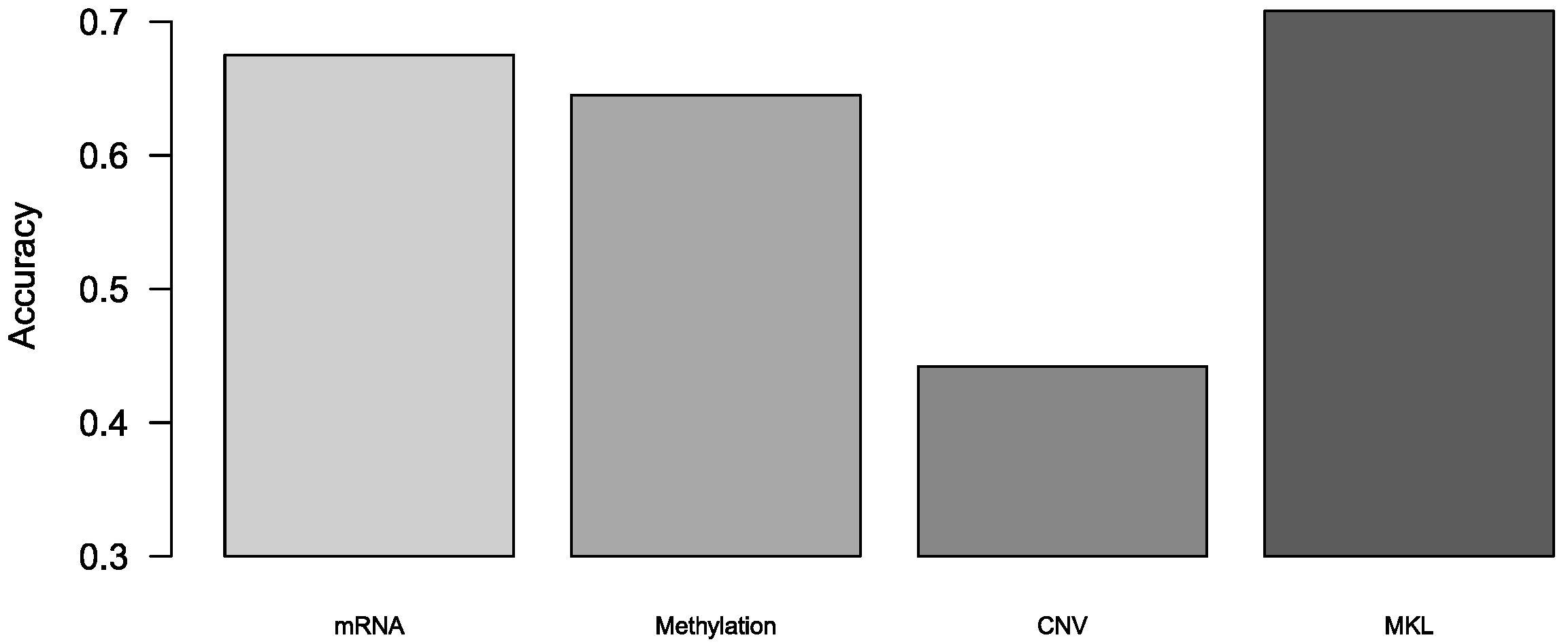
3.1. Comparison Binary Classification by Different Omics
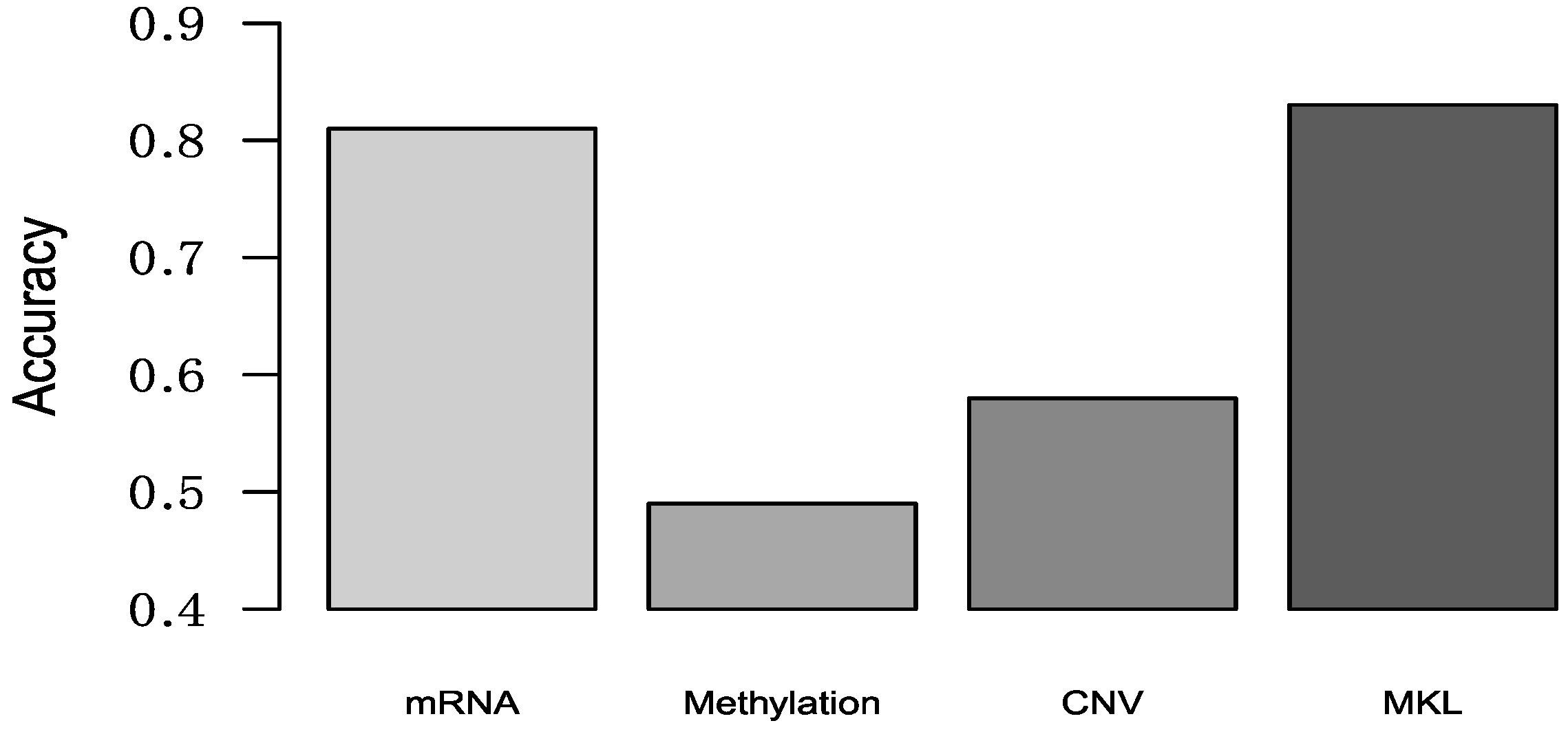
3.2. Comparison Multi-Classification by Different Omics
3.3. Comparison Subtypes of Triple-Negative Breast Cancer Multi-Classification
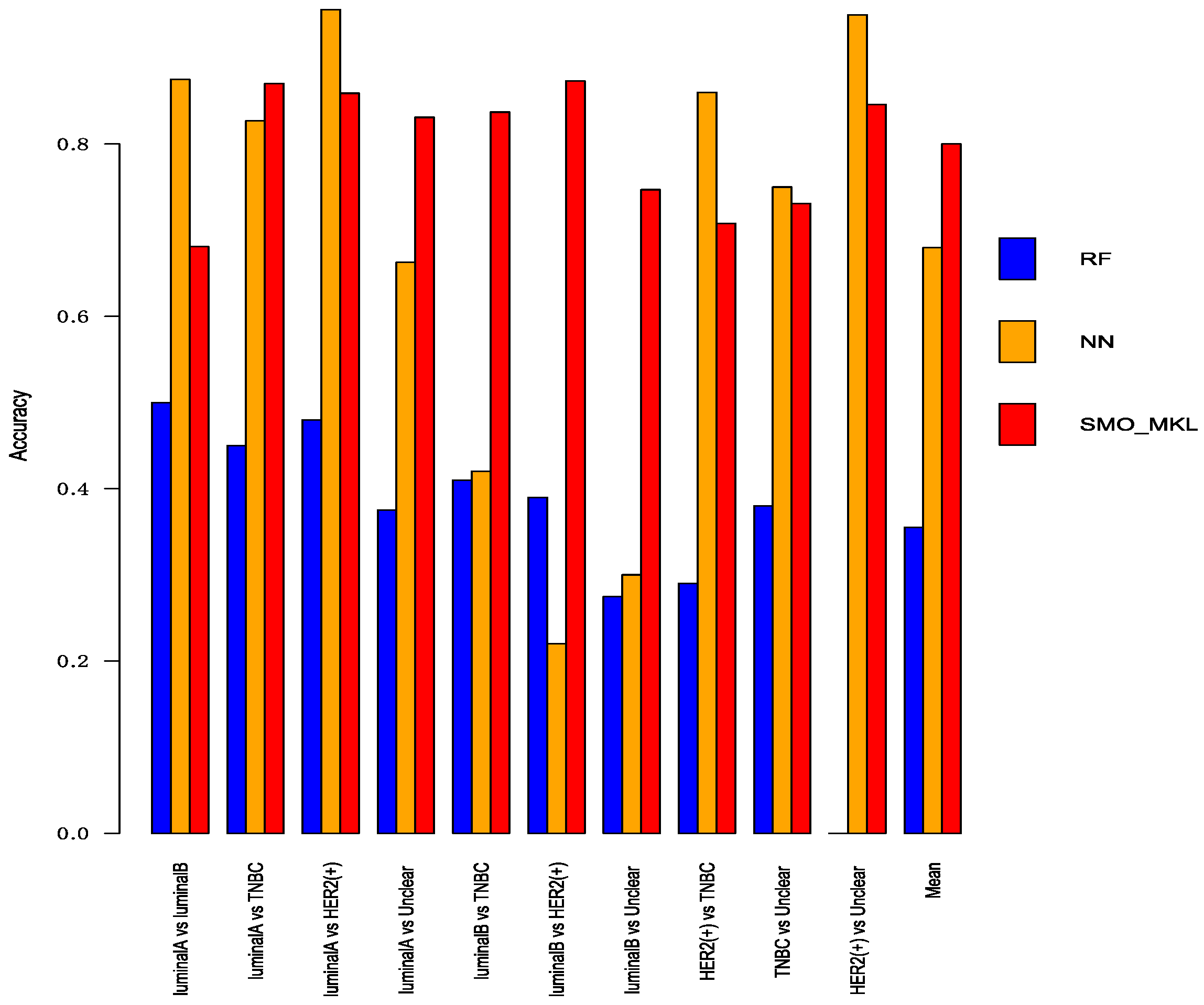
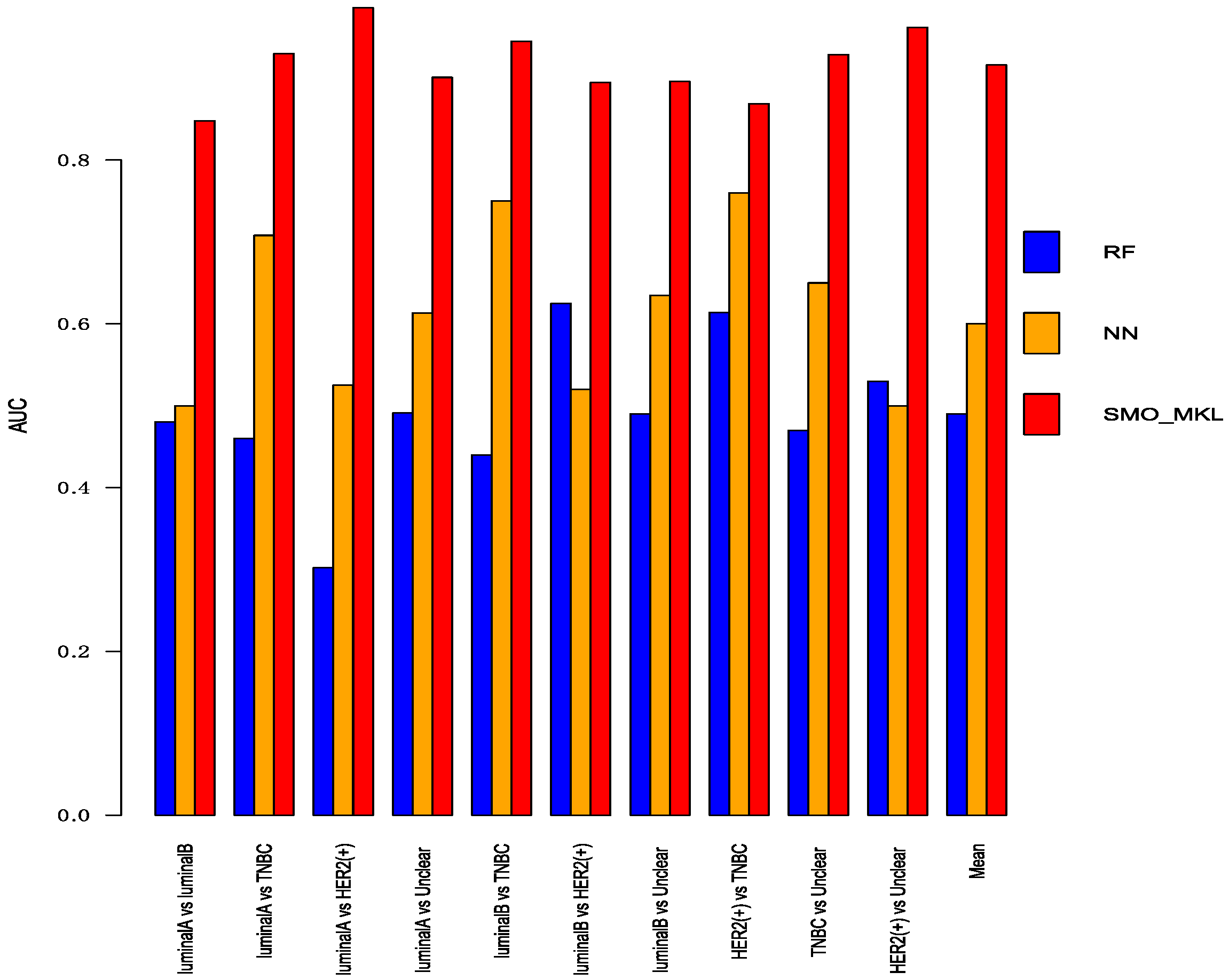
3.4. Comparison with Other Methods
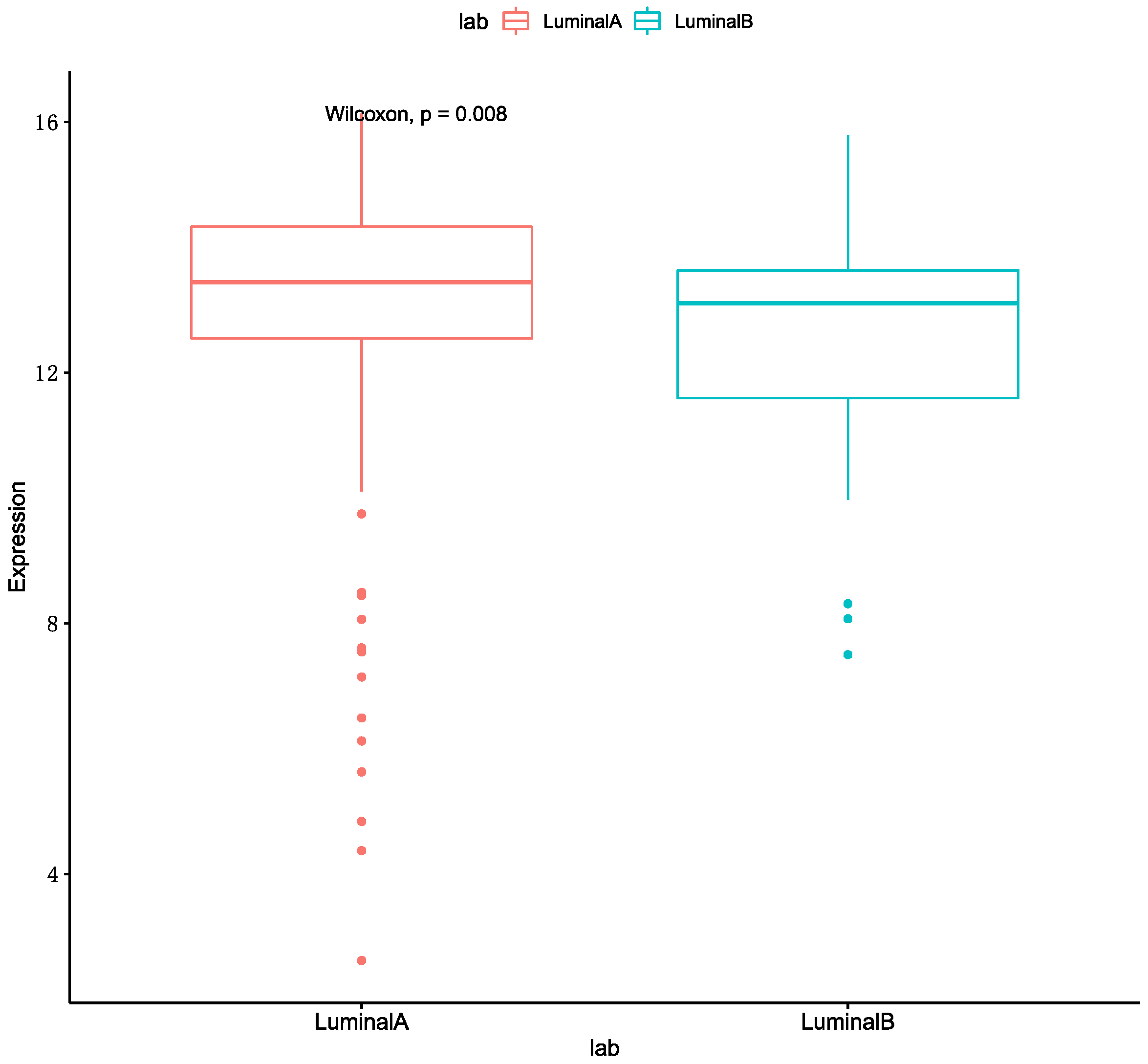
3.5. Analysis of Selected Genes
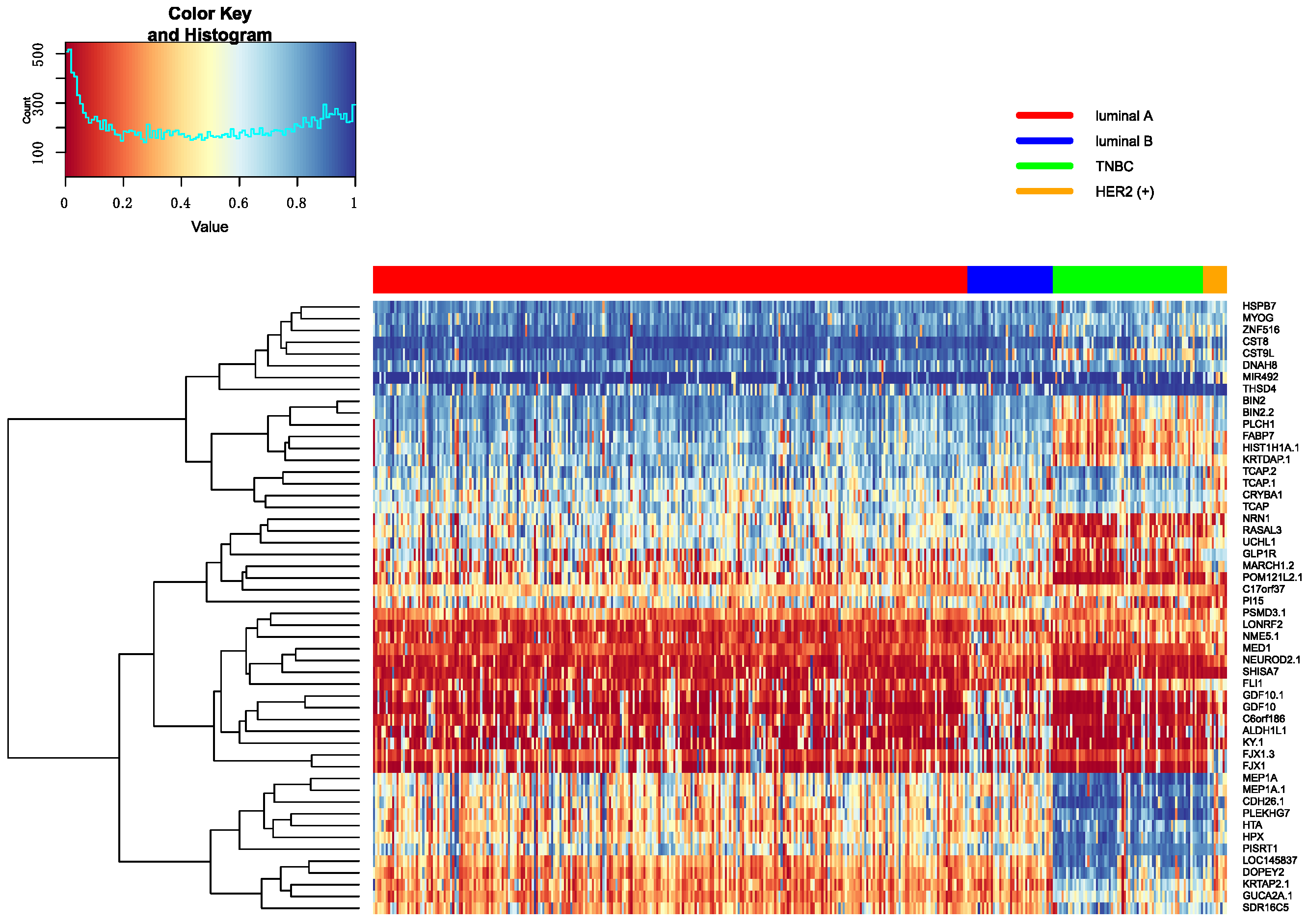
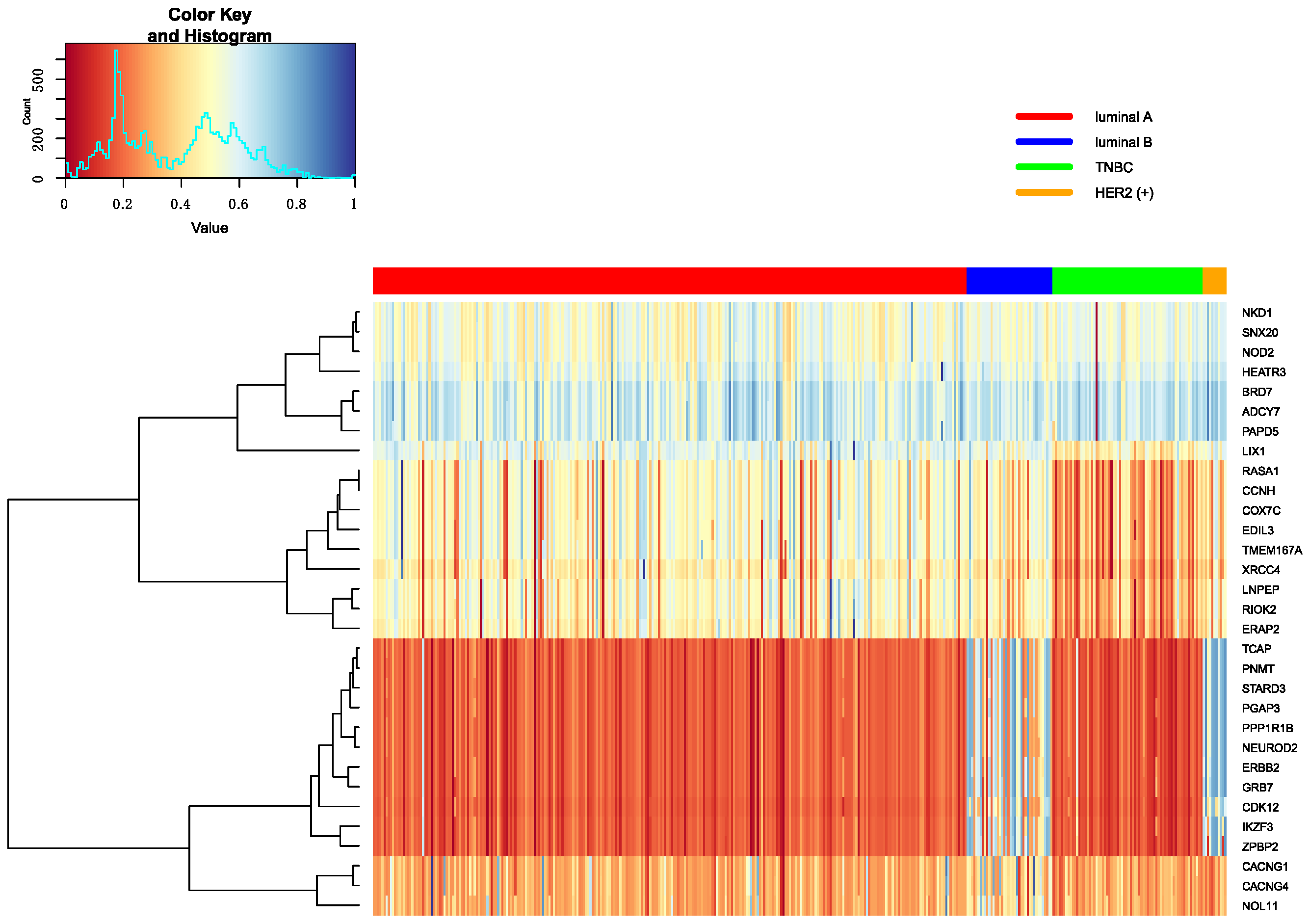
3.5.1. Heatmap of Selected Genes
3.5.2. Enrichment of Selected Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Callahan, R.; Hurvitz, S. HER2-Positive Breast Cancer: Current Management of Early, Advanced, and Recurrent Disease. Curr. Opin. Obstet. Gynecol. 2011, 23, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Assi, H.A.; Khoury, K.E.; Dbouk, H.; Khalil, L.E.; Mouhieddine, T.H.; El Saghir, N.S. Epidemiology and prognosis of breast cancer in young women. J. Thorac. Dis. 2013, 5, S2–S8. [Google Scholar] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of Breast Cancer by Immunohistochemistry to Investigate a Relationship between Subtype and Short and Long Term Survival: A Collaborative Analysis of Data for 10,159 Cases from 12 Studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.L.; Taghian, A.G.; Katz, M.S.; Niemierko, A.; Abi Raad, R.F.; Boon, W.L.; Bellon, J.R.; Wong, J.S.; Smith, B.L.; Harris, J.R. Breast cancer subtype approximated by estrogen receptor, progesterone receptor, and HER-2 is associated with local and distant recurrence after breast-conserving therapy. J. Clin. Oncol. 2008, 26, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Inic, Z.; Zegarac, M.; Inic, M.; Markovic, I.; Kozomara, Z.; Djurisic, I.; Inic, I.; Pupic, G.; Jancic, S. Difference between Luminal A and Luminal B Subtypes According to Ki-67, Tumor Size, and Progesterone Receptor Negativity Providing Prognostic Information. Clin. Med. Insights Oncol. 2014, 8, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Ades, F.; Zardavas, D.; Bozovic-Spasojevic, I.; Pugliano, L.; Fumagalli, D.; de Azambuja, E.; Viale, G.; Sotiriou, C.; Piccart, M. Luminal B Breast Cancer: Molecular Characterization, Clinical Management, and Future Perspectives. J. Clin. Oncol. 2014, 32, 2794–2803. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jiang, Y.Z.; Hu, X.; Sun, W.; Liu, Y.R.; Liu, F.; Luo, R.C.; Shao, Z.M. Clinicopathological characteristics of patients with HER2-positive breast cancer and the efficacy of trastuzumab in the People’s Republic of China. Oncol. Targets Ther. 2010, 9, 2287–2295. [Google Scholar]
- De Laurentiis, M.; Cianniello, D.; Caputo, R.; Stanzione, B.; Arpino, G.; Cinieri, S.; Lorusso, V.; De Placido, S. Treatment of triple negative breast cancer (TNBC): Current options and future perspectives. Cancer Treat Rev. 2010, 36, S80–S86. [Google Scholar] [CrossRef]
- Plasilova, M.L.; Hayse, B.; Killelea, B.K.; Horowitz, N.R.; Chagpar, A.B.; Lannin, D.R. Features of triple-negative breast cancer. Medicine 2016, 95, e4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10898. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Horlings, H.M.; Kreike, B.; Hayes, M.M.; Hauptmann, M.; Wessels, L.F.; de Jong, D.; Van de Vijver, M.J.; Van’t Veer, L.J.; Peterse, J.L. Refinement of breast cancer classification by molecular characterization of histological special types. J. Pathol. 2008, 216, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Liang, X.; Xu, R. A Multiple Kernel Learning Model Based on p-Norm. Comput. Intell. Neurosci. 2018, 2018, 1018789. [Google Scholar] [CrossRef] [PubMed]
- Mehmet, G.; Ethem, A. Multiple Kernel Learning Algorithms. J. Mach. Learn. Res. 2011, 12, 2211–2268. [Google Scholar]
- Song, T.; Wang, Y.; Du, W.; Cao, S.; Tian, Y.; Liang, Y. The method for breast cancer grade prediction and pathway analysis based on improved multiple kernel learning. J. Bioinf. Comput. Biol. 2017, 15, 1650037. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Cao, Z.; Song, T.; Li, Y.; Liang, Y. A feature selection method based on multiple kernel learning with expression profiles of different types. BioData Min. 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashmi, A.A.; Aijaz, S.; Khan, S.M.; Mahboob, R.; Irfan, M.; Zafar, N.I.; Nisar, M.; Siddiqui, M.; Edhi, M.M.; Faridi, N.; et al. Prognostic parameters of luminal A and luminal B intrinsic breast cancer subtypes of Pakistani patients. World J. Surg. Oncol. 2018, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.H.; Man, H.T.; Zhao, X.D.; Dong, N.; Ma, S.L. Estrogen receptor-positive breast cancer molecular signatures and therapeutic potentials. Biomed. Rep. 2014, 2, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Gray, W.H.; Lemann, B.D.; Bauer, J.A.; Shyr, Y.; Pietenpol, J.A. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inf. 2012, 11, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Klein-Seetharaman, J.; Bar-Joseph, Z. Random Forest Similarity for Protein-Protein Interaction Prediction from Multiple Sources. Pac. Symp. Biocomput. 2005, 2005, 531–542. [Google Scholar]
- Cui, J.; Germer, K.; Wu, T.; Wang, J.; Luo, J.; Wang, S.C.; Wang, Q.; Zhang, X. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer Res. 2012, 72, 5625–5634. [Google Scholar] [CrossRef] [PubMed]
- Linda, L.; Nancy, Y.; Adina, I.; Christina, Y.; van’t Veer, L.; Nordenskjöld, B.; Benz, C.; Fornander, T.; Stål, O.; Czene, K.; et al. Long-term benefit from tamoxifen therapy for patients with Luminal A and Luminal B breast cancer: Retrospective analysis of the STO-3 trial. J. Clin. Oncol. 2018, 15, 541. [Google Scholar]
- Shaoxiao, T.; Baohua, Y.; Xiaoli, X.; Yufan, C.; Xiaoyun, T.; Hongfen, L.; Rui, B.; Xiangjie, S.; Ruohong, S.; Wentao, Y. Characterisation of GATA3 expression in invasive breast cancer: Difference in histological subtypes and immunohistochemically defined molecular subtypes. J. Clin Pathol. 2017, 70, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Varambally, S.; Ding, L.; Shen, R.; Sabel, M.S.; Ghosh, D.; Chinnaiyan, A.M.; Kleer, C.G. Identification of GATA3 as a Breast Cancer Prognostic Marker by Global Gene Expression Meta-analysis. Cancer Res. 2015, 65, 11259–11264. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.J.; Stocco, D.M. Cholesterol Transporters of the START Domain Protein Family in Health and Disease; Springer: New York, NY, USA, 2014; pp. 129–134. [Google Scholar]
- Tyanova, S.; Albrechtsen, R.; Krongvist, P.; Cox, J.; Mann, M.; Geiger, T. Proteomic maps of breast cancer type. Nat. Commun. 2016, 7, 10259. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.M.; Camps, C.; Winchester, L.; Snell, C.E.; Gee, H.E.; Sheldon, H.; Taylor, M.; Harris, A.L.; Ragoussis, J. MicroRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 2011, 71, 5635–5645. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Chen, A.; Bai, Z. Integrative investigation on breast cancer in ER, PR and HER2-defined subgroups using mRNA and miRNA expression profilin. Sci. Rep. 2014, 4, 6566. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Zhu, H.; Bi, S.J. Pathological features and prognosis of different molecular subtypes of breast cancer. Mol. Med. Rep. 2012, 6, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- McDonald, E.S.; Clark, A.S.; Tchou, J.; Zhang, P.; Freedman, G.M. Clinical Diagnosis and Management of Breast Cancer. J. Nucl. Med. 2016, 27, 9S–16S. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breas. 2015, 24, S26–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kom, M.; DelLisi, C. Pathway-based classification of breast cancer subtypes. Biol. Direct. 2012, 22, 1697–1712. [Google Scholar]
- Wu, T.; Wang, T.; Jiang, R.; Lu, X.; Tian, J. A pathways-based prediction model for classifying breast cancer subtypes. Oncotarget 2017, 8, 58809–58822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tang, X.Q.; Bai, Z.; Dai, X. Exploring the intrinsic differences among breast tumor subtypes defined using immunohistochemistry markers based on the decision tree. Sci. Rep. 2016, 6, 35773. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Subtypes | Definition |
|---|---|
| Luminal A | ER/PR+, Her2− |
| Luminal B | ER/PR+, Her2+ |
| TNBC | ER/PR−, Her2− |
| HER2 (+) | ER/PR−, Her2+ |
| Unclear | Other samples |
| Breast Subtypes | Cancer Patients |
|---|---|
| Luminal A | 277 |
| Luminal B | 40 |
| TNBC | 70 |
| HER2 (+) | 11 |
| Unclear | 208 |
| Breast Cancer Subtypes | mRNA | Methylation | CNV | MKL |
|---|---|---|---|---|
| Luminal A vs. luminal B | 0.436 | 0.436 | 0.490 | 0.681 |
| Luminal A vs. HER2 (+) | 0.739 | 0.566 | 0.739 | 0.870 |
| Luminal A vs. TNBC | 0.868 | 0.867 | 0.604 | 0.859 |
| Luminal A vs. Unclear | 0.760 | 0.849 | 0.473 | 0.831 |
| Luminal B vs. HER2 (+) | 0.732 | 0.776 | 0.485 | 0.837 |
| Luminal B vs. TNBC | 0.871 | 0.883 | 0.855 | 0.873 |
| Luminal B vs. Unclear | 0.696 | 0.748 | 0.770 | 0.747 |
| HER2 (+) vs. TNBC | 0.5 | 0.5 | 0.5 | 0.708 |
| HER2 (+) vs. Unclear | 0.495 | 0.498 | 0.5 | 0.731 |
| TNBC vs. Unclear | 0.806 | 0.836 | 0.717 | 0.846 |
| Mean | 0.690 | 0.696 | 0.613 | 0.798 |
| Breast Cancer Subtypes | mRNA | Methylation | CNV | MKL |
|---|---|---|---|---|
| Luminal A vs. luminal B | 0.835 | 0.632 | 0.810 | 0.848 |
| Luminal A vs. HER2 (+) | 0.973 | 0.903 | 0.979 | 0.986 |
| Luminal A vs. TNBC | 0.934 | 0.926 | 0.909 | 0.930 |
| Luminal A vs. Unclear | 0.824 | 0.878 | 0.589 | 0.901 |
| Luminal B vs. HER2 (+) | 0.843 | 0.824 | 0.725 | 0.895 |
| Luminal B vs. TNBC | 0.947 | 0.932 | 0.941 | 0.945 |
| Luminal B vs. Unclear | 0.875 | 0.808 | 0.835 | 0.896 |
| HER2 (+) vs. TNBC | 0.867 | 0.778 | 0.741 | 0.869 |
| HER2 (+) vs. Unclear | 0.925 | 0.873 | 0.859 | 0.962 |
| TNBC vs. Unclear | 0.902 | 0.918 | 0.834 | 0.929 |
| Mean | 0.893 | 0.847 | 0.822 | 0.916 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, M.; Song, T.; Du, W.; Han, S.; Zuo, C.; Li, Y.; Wang, Y.; Yang, Z. Classifying Breast Cancer Subtypes Using Multiple Kernel Learning Based on Omics Data. Genes 2019, 10, 200. https://doi.org/10.3390/genes10030200
Tao M, Song T, Du W, Han S, Zuo C, Li Y, Wang Y, Yang Z. Classifying Breast Cancer Subtypes Using Multiple Kernel Learning Based on Omics Data. Genes. 2019; 10(3):200. https://doi.org/10.3390/genes10030200
Chicago/Turabian StyleTao, Mingxin, Tianci Song, Wei Du, Siyu Han, Chunman Zuo, Ying Li, Yan Wang, and Zekun Yang. 2019. "Classifying Breast Cancer Subtypes Using Multiple Kernel Learning Based on Omics Data" Genes 10, no. 3: 200. https://doi.org/10.3390/genes10030200
APA StyleTao, M., Song, T., Du, W., Han, S., Zuo, C., Li, Y., Wang, Y., & Yang, Z. (2019). Classifying Breast Cancer Subtypes Using Multiple Kernel Learning Based on Omics Data. Genes, 10(3), 200. https://doi.org/10.3390/genes10030200