DNA Methylation Reprogramming during Mammalian Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNA Methyltransferases and Regulators
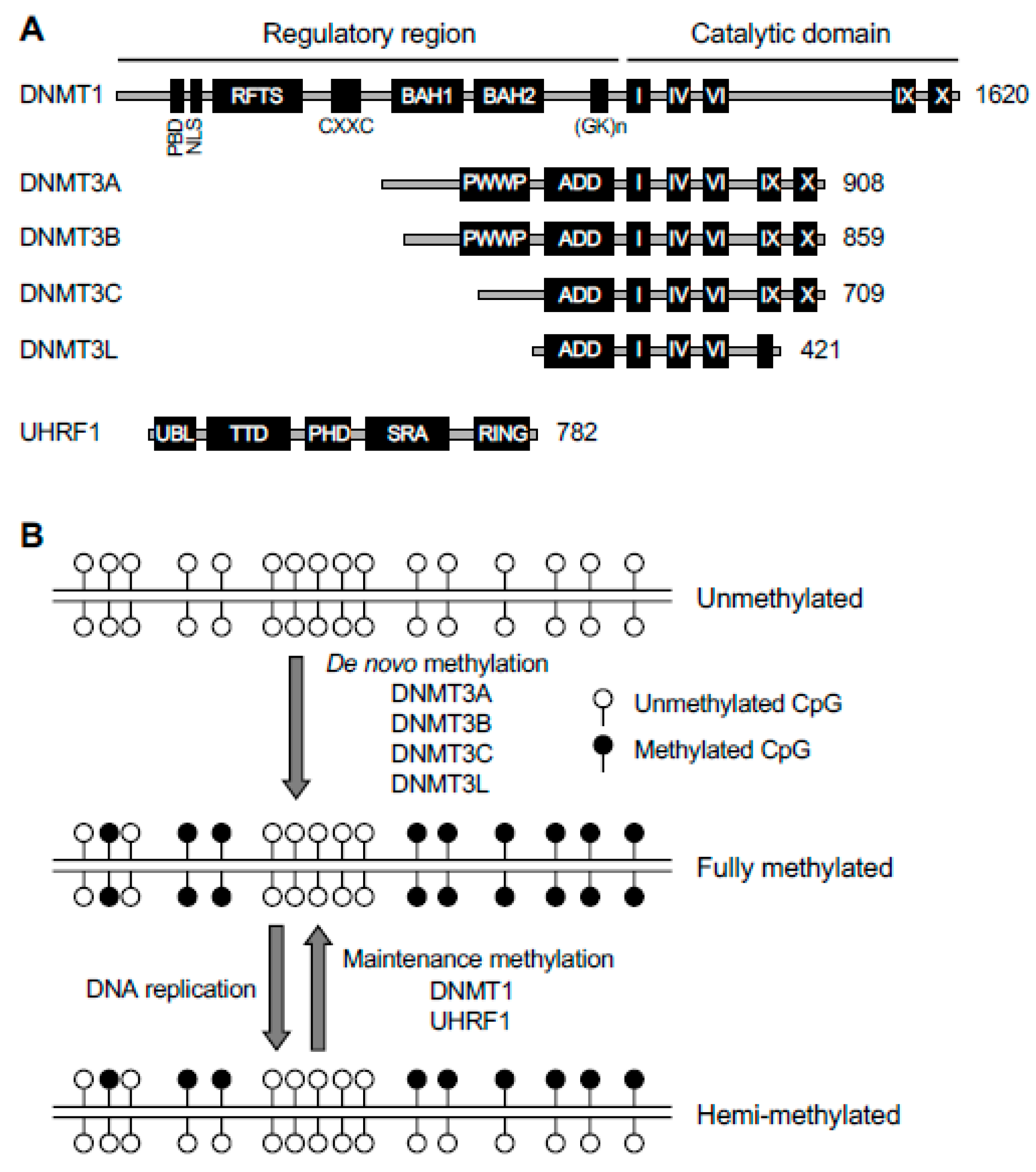
2.1. DNMT1 and UHRF1—Key Components of Maintenance Methylation Machinery
2.2. DNMT3A, DNMT3B, DNMT3C, and DNMT3L—Key Components of de novo Methylation Machinery
2.3. DNMT2/TRDMT1
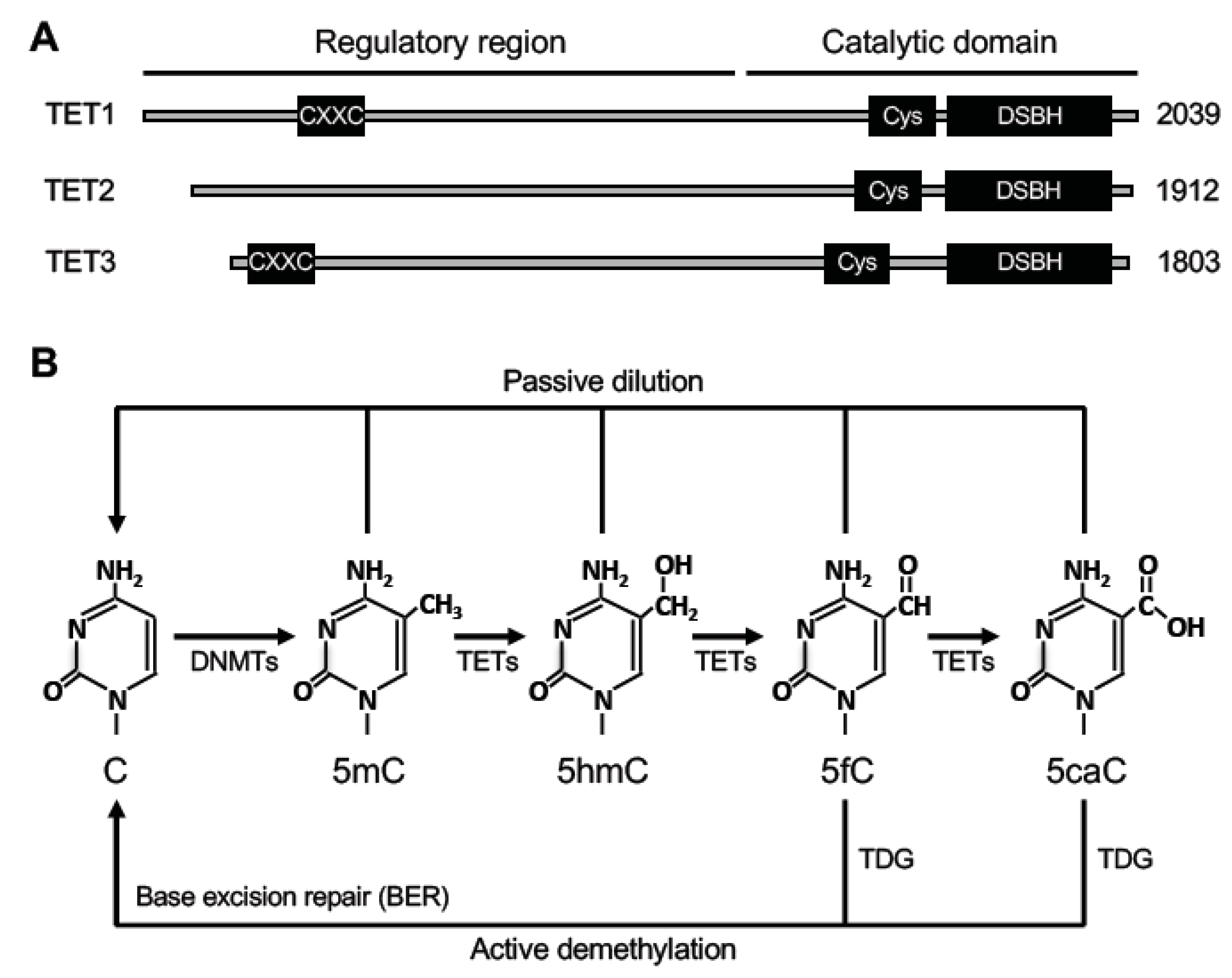
3. DNA Demethylation
3.1. Passive DNA Demethylation
3.2. Active DNA Demethylation
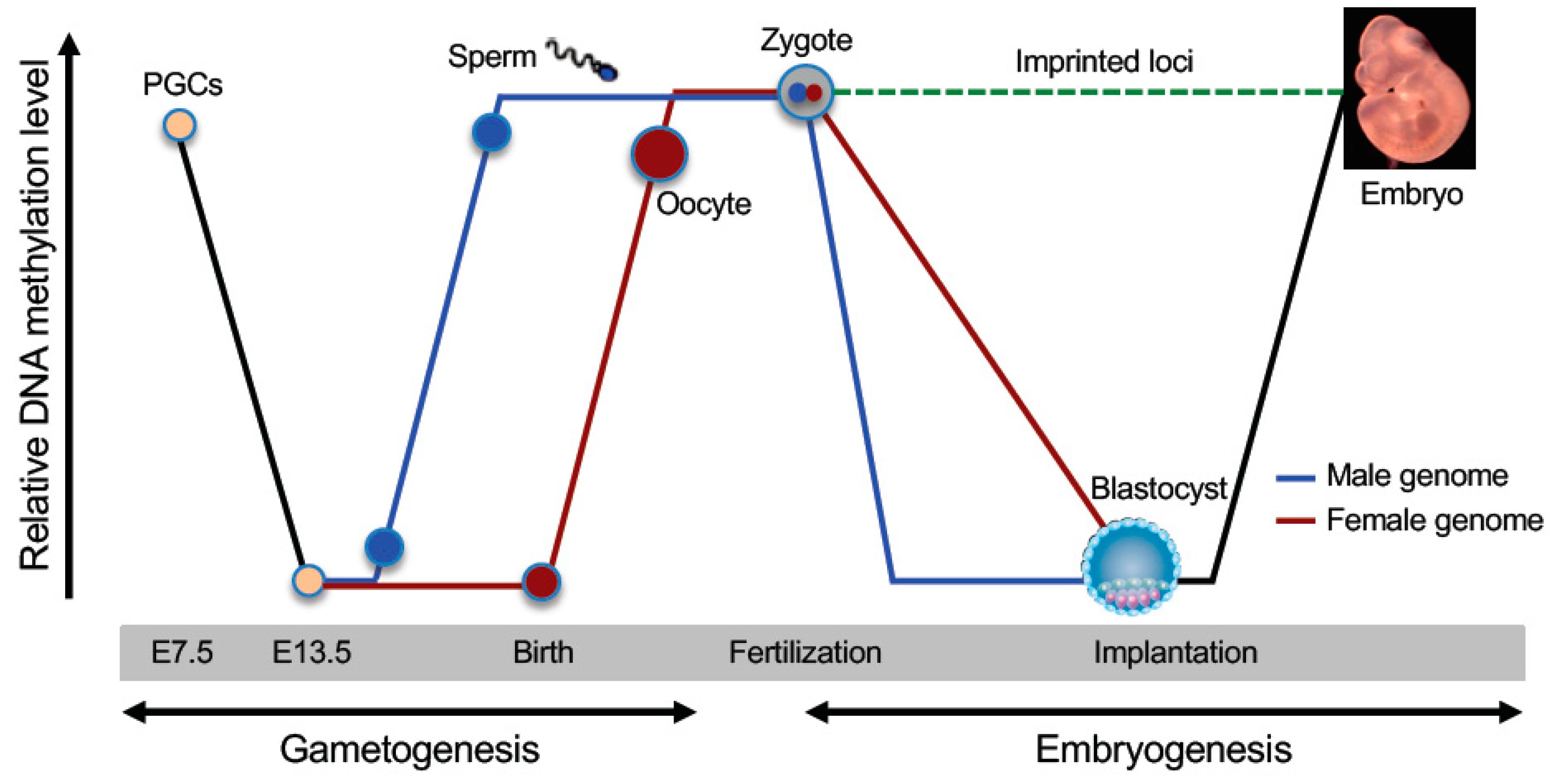
4. DNA Methylation Reprogramming in the Germline
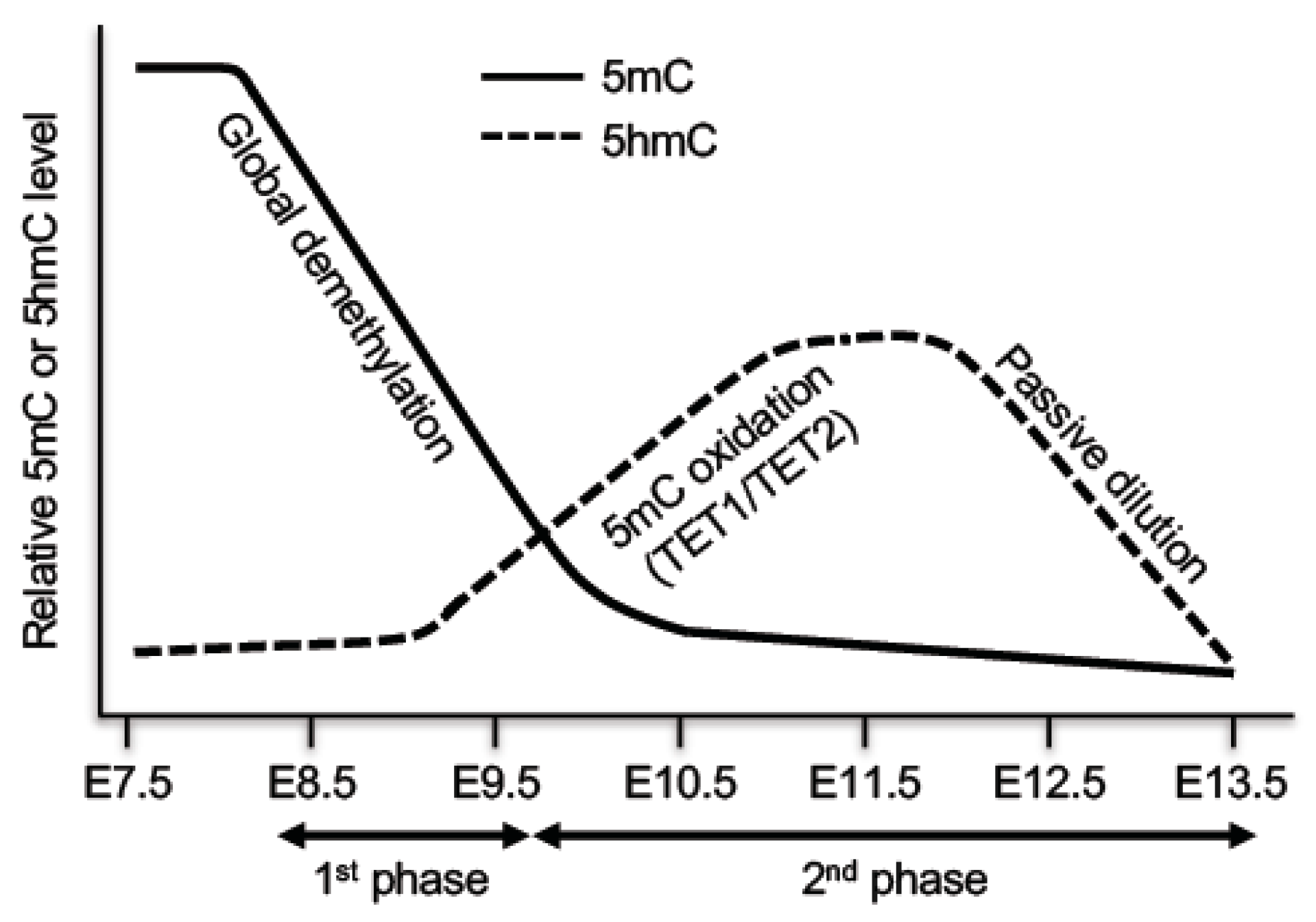
4.1. Biphasic Demethylation in PGCs
4.2. Sex-Specific Remethylation in the Germline
4.3. Establishment of Methylation Imprints
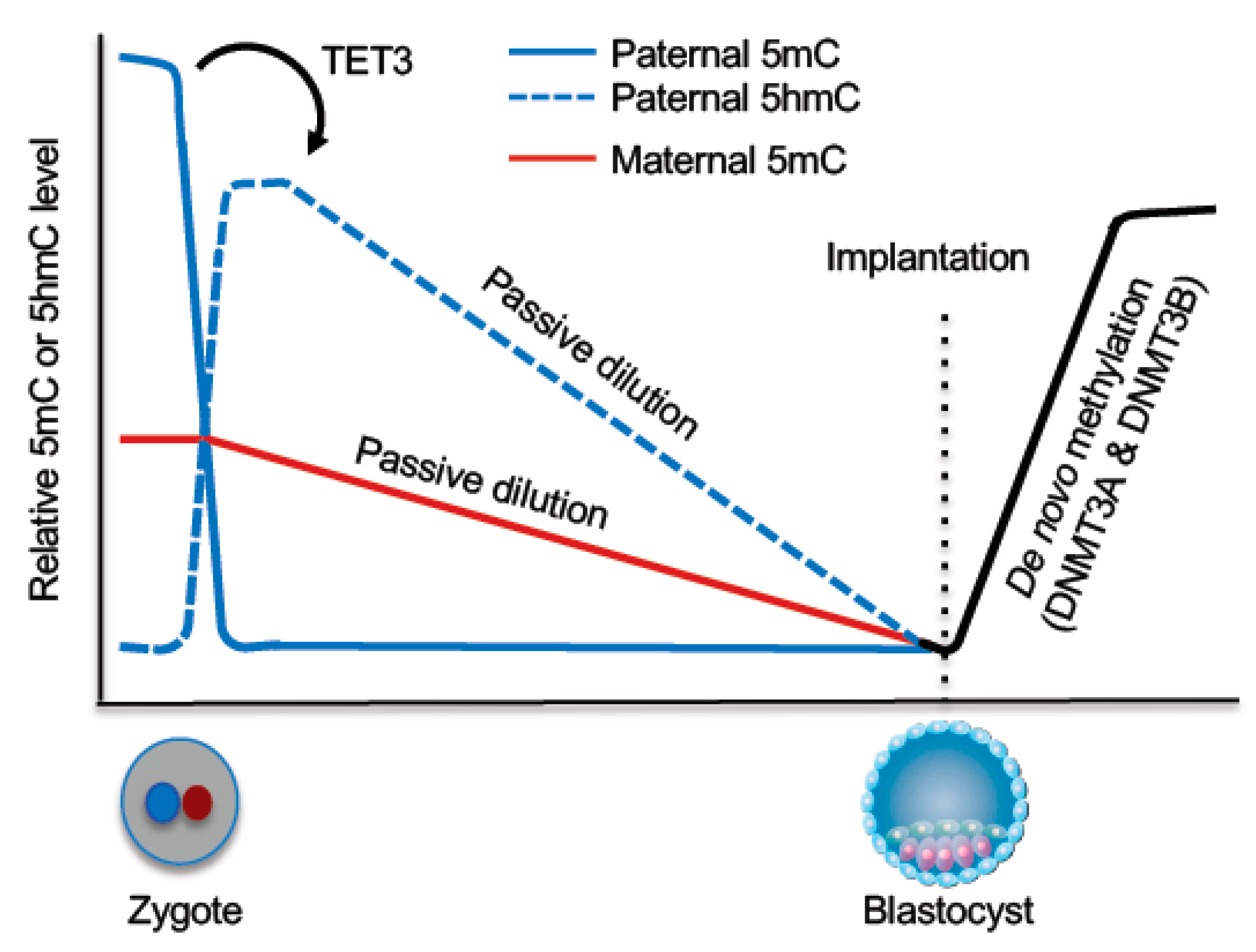
5. DNA Methylation Reprogramming in Early Embryos
5.1. DNA Demethylation During Preimplantation Development
5.2. Maintenance of Methylation Imprints
5.3. De novo Methylation in the Epiblast
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, T.; Singh, A.K.; Chen, T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015, 7, 247–265. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold. Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Kelsey, G. De novo DNA methylation: a germ cell perspective. Trends Genet 2012, 28, 33–42. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Chen, T.; Li, E. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 2004, 60, 55–89. [Google Scholar] [PubMed]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988, 203, 971–983. [Google Scholar] [CrossRef]
- Mertineit, C.; Yoder, J.A.; Taketo, T.; Laird, D.W.; Trasler, J.M.; Bestor, T.H. Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development 1998, 125, 889–897. [Google Scholar]
- Rouleau, J.; Tanigawa, G.; Szyf, M. The mouse DNA methyltransferase 5’-region. A unique housekeeping gene promoter. J. Biol. Chem. 1992, 267, 7368–7377. [Google Scholar]
- Ding, F.; Chaillet, J.R. In vivo stabilization of the Dnmt1 (cytosine-5)- methyltransferase protein. Proc. Natl. Acad. Sci. USA 2002, 99, 14861–14866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, K.; Suetake, I.; Yamashita, E.; Suga, M.; Narita, H.; Nakagawa, A.; Tajima, S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. USA 2011, 108, 9055–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, S.; Murata, T.; Ugai, H.; Yamazaki, T.; Yokoyama, K.K. Control elements of Dnmt1 gene are regulated in cell-cycle dependent manner. Nucleic Acids Res. Suppl. 2003, 3, 307–308. [Google Scholar] [CrossRef]
- Leonhardt, H.; Page, A.W.; Weier, H.U.; Bestor, T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Lei, H.; Oh, S.P.; Okano, M.; Juttermann, R.; Goss, K.A.; Jaenisch, R.; Li, E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1996, 122, 3195–3205. [Google Scholar] [PubMed]
- Li, Y.; Zhang, Z.; Chen, J.; Liu, W.; Lai, W.; Liu, B.; Li, X.; Liu, L.; Xu, S.; Dong, Q.; et al. Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature 2018, 564, 136–140. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Q.; Li, P.; Zhao, Q.; Zhang, J.; Li, J.; Koseki, H.; Wong, J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013, 4, 1563. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Krajewski, K.; Nady, N.; Tempel, W.; Xue, S.; Badeaux, A.I.; Barsyte-Lovejoy, D.; Martinez, J.Y.; Bedford, M.T.; Fuchs, S.M.; et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012, 19, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Dickson, B.M.; Ong, M.S.; Krajewski, K.; Houliston, S.; Kireev, D.B.; Arrowsmith, C.H.; Strahl, B.D. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013, 27, 1288–1298. [Google Scholar] [CrossRef] [Green Version]
- Rajakumara, E.; Wang, Z.; Ma, H.; Hu, L.; Chen, H.; Lin, Y.; Guo, R.; Wu, F.; Li, H.; Lan, F.; et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell 2011, 43, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, Z.; Wang, P.; Lin, Y.; Xu, Y. Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res. 2011, 21, 1374–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Shen, J.; Yang, Z.; Chen, P.; Zhao, B.; Hu, W.; Lan, W.; Tong, X.; Wu, H.; Li, G.; et al. Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res. 2011, 21, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Yamaguchi, L.; Sharif, J.; Johmura, Y.; Kawamura, T.; Nakanishi, K.; Shimamura, S.; Arita, K.; Kodama, T.; Ishikawa, F.; et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013, 502, 249–253. [Google Scholar] [CrossRef]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forne, I.; Pichler, G.; Horl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, J.S.; Cornett, E.M.; Goldfarb, D.; DaRosa, P.A.; Li, Z.M.; Yan, F.; Dickson, B.M.; Guo, A.H.; Cantu, D.V.; Kaustov, L.; et al. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. Elife 2016, 5, e17101. [Google Scholar] [CrossRef]
- Fang, J.; Cheng, J.; Wang, J.; Zhang, Q.; Liu, M.; Gong, R.; Wang, P.; Zhang, X.; Feng, Y.; Lan, W.; et al. Hemi-methylated DNA opens a closed conformation of UHRF1 to facilitate its histone recognition. Nat. Commun. 2016, 7, 11197. [Google Scholar] [CrossRef] [Green Version]
- Foster, B.M.; Stolz, P.; Mulholland, C.B.; Montoya, A.; Kramer, H.; Bultmann, S.; Bartke, T. Critical Role of the UBL Domain in Stimulating the E3 Ubiquitin Ligase Activity of UHRF1 toward Chromatin. Mol. Cell 2018, 72, 739–752. [Google Scholar] [CrossRef] [PubMed]
- DaRosa, P.A.; Harrison, J.S.; Zelter, A.; Davis, T.N.; Brzovic, P.; Kuhlman, B.; Klevit, R.E. A Bifunctional Role for the UHRF1 UBL Domain in the Control of Hemi-methylated DNA-Dependent Histone Ubiquitylation. Mol. Cell 2018, 72, 753–765. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ueda, Y.; Xie, S.; Li, E. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 2002, 277, 38746–38754. [Google Scholar] [CrossRef] [PubMed]
- Ostler, K.R.; Yang, Q.; Looney, T.J.; Zhang, L.; Vasanthakumar, A.; Tian, Y.; Kocherginsky, M.; Raimondi, S.L.; DeMaio, J.G.; Salwen, H.R.; et al. Truncated DNMT3B isoform DNMT3B7 suppresses growth, induces differentiation, and alters DNA methylation in human neuroblastoma. Cancer Res. 2012, 72, 4714–4723. [Google Scholar] [CrossRef] [Green Version]
- Duymich, C.E.; Charlet, J.; Yang, X.; Jones, P.A.; Liang, G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat. Commun. 2016, 7, 11453. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Sawada, K.; Zhang, X.; Cheng, X. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 2002, 9, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Tsujimoto, N.; Li, E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 2004, 24, 9048–9058. [Google Scholar] [CrossRef]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schubeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef]
- Rondelet, G.; Dal Maso, T.; Willems, L.; Wouters, J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 2016, 194, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, J.; Nankumo, T.; Arita, K.; Inamoto, S.; Ariyoshi, M.; Shirakawa, M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009, 10, 1235–1241. [Google Scholar] [CrossRef]
- Guo, X.; Wang, L.; Li, J.; Ding, Z.; Xiao, J.; Yin, X.; He, S.; Shi, P.; Dong, L.; Li, G.; et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 2015, 517, 640–644. [Google Scholar] [CrossRef]
- Watanabe, D.; Suetake, I.; Tada, T.; Tajima, S. Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis. Mech. Dev. 2002, 118, 187–190. [Google Scholar] [CrossRef]
- Sakai, Y.; Suetake, I.; Shinozaki, F.; Yamashina, S.; Tajima, S. Co-expression of de novo DNA methyltransferases Dnmt3a2 and Dnmt3L in gonocytes of mouse embryos. Gene Expr. Patterns 2004, 5, 231–237. [Google Scholar] [CrossRef]
- Ma, P.; de Waal, E.; Weaver, J.R.; Bartolomei, M.S.; Schultz, R.M. A DNMT3A2-HDAC2 Complex Is Essential for Genomic Imprinting and Genome Integrity in Mouse Oocytes. Cell Rep. 2015, 13, 1552–1560. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed]
- Lees-Murdock, D.J.; McLoughlin, G.A.; McDaid, J.R.; Quinn, L.M.; O’Doherty, A.; Hiripi, L.; Hack, C.J.; Walsh, C.P. Identification of 11 pseudogenes in the DNA methyltransferase gene family in rodents and humans and implications for the functional loci. Genomics 2004, 84, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Barau, J.; Teissandier, A.; Zamudio, N.; Roy, S.; Nalesso, V.; Herault, Y.; Guillou, F.; Bourc’his, D. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 2016, 354, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Kawasaki, K.; Scott, H.S.; Ollila, J.; Vihinen, M.; Heino, M.; Shintani, A.; Minoshima, S.; Krohn, K.; Antonarakis, S.E.; et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000, 65, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Lyle, R.; Krohn, K.; Antonarakis, S.E.; Peterson, P. Isolation and initial characterization of the mouse Dnmt3l gene. Cytogenet. Cell Genet. 2001, 92, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002, 129, 1983–1993. [Google Scholar] [PubMed]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef] [Green Version]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef] [PubMed]
- Veland, N.; Lu, Y.; Hardikar, S.; Gaddis, S.; Zeng, Y.; Liu, B.; Estecio, M.R.; Takata, Y.; Lin, K.; Tomida, M.W.; et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 2019, 47, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourc’his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96–99. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Tomizawa, S.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811–814. [Google Scholar] [CrossRef] [Green Version]
- Shirane, K.; Toh, H.; Kobayashi, H.; Miura, F.; Chiba, H.; Ito, T.; Kono, T.; Sasaki, H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013, 9, e1003439. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Krepelova, A.; Incarnato, D.; Maldotti, M.; Parlato, C.; Galvagni, F.; Matarese, F.; Stunnenberg, H.G.; Oliviero, S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 2013, 155, 121–134. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Res. 1998, 26, 2536–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Shi, J.; Tuorto, F.; Li, X.; Liu, Y.; Liebers, R.; Zhang, L.; Qu, Y.; Qian, J.; et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat. Cell Biol. 2018, 20, 535–540. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef]
- Kagiwada, S.; Kurimoto, K.; Hirota, T.; Yamaji, M.; Saitou, M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013, 32, 340–353. [Google Scholar] [CrossRef]
- Dan, J.; Rousseau, P.; Hardikar, S.; Veland, N.; Wong, J.; Autexier, C.; Chen, T. Zscan4 Inhibits Maintenance DNA Methylation to Facilitate Telomere Elongation in Mouse Embryonic Stem Cells. Cell Rep. 2017, 20, 1936–1949. [Google Scholar] [CrossRef] [PubMed]
- Veland, N.; Hardikar, S.; Zhong, Y.; Gayatri, S.; Dan, J.; Strahl, B.D.; Rothbart, S.B.; Bedford, M.T.; Chen, T. The Arginine Methyltransferase PRMT6 Regulates DNA Methylation and Contributes to Global DNA Hypomethylation in Cancer. Cell Rep. 2017, 21, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lu, J.; Cheng, J.; Rao, Q.; Li, Z.; Hou, H.; Lou, Z.; Zhang, L.; Li, W.; Gong, W.; et al. Structural insight into substrate preference for TET-mediated oxidation. Nature 2015, 527, 118–122. [Google Scholar] [CrossRef]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Aijo, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lahdesmaki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013. [Google Scholar] [CrossRef]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012, 40, 4841–4849. [Google Scholar] [CrossRef] [Green Version]
- Saitou, M. Germ cell specification in mice. Curr. Opin. Genet. Dev. 2009, 19, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Guibert, S.; Forne, T.; Weber, M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012, 22, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seisenberger, S.; Andrews, S.; Krueger, F.; Arand, J.; Walter, J.; Santos, F.; Popp, C.; Thienpont, B.; Dean, W.; Reik, W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell 2012, 48, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Hong, K.; Liu, R.; Inoue, A.; Shen, L.; Zhang, K.; Zhang, Y. Dynamics of 5-methylcytosine and 5-hydroxymethylcytosine during germ cell reprogramming. Cell Res. 2013, 23, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.J.; Huang, Y.; Chen, P.Y.; Feng, S.; Calvopina, J.H.; Nee, K.; Lee, S.A.; Le, T.; Yoon, A.J.; Faull, K.; et al. Stage-specific roles for Tet1 and Tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell 2013, 12, 470–478. [Google Scholar] [CrossRef]
- Piccolo, F.M.; Bagci, H.; Brown, K.E.; Landeira, D.; Soza-Ried, J.; Feytout, A.; Mooijman, D.; Hajkova, P.; Leitch, H.G.; Tada, T.; et al. Different Roles for Tet1 and Tet2 Proteins in Reprogramming-Mediated Erasure of Imprints Induced by EGC Fusion. Mol. Cell 2013, 49, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Hong, K.; Liu, R.; Shen, L.; Inoue, A.; Diep, D.; Zhang, K.; Zhang, Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature 2012, 492, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Shen, L.; Liu, Y.; Sendler, D.; Zhang, Y. Role of Tet1 in erasure of genomic imprinting. Nature 2013, 504, 460–464. [Google Scholar] [CrossRef]
- Hill, P.W.S.; Leitch, H.G.; Requena, C.E.; Sun, Z.; Amouroux, R.; Roman-Trufero, M.; Borkowska, M.; Terragni, J.; Vaisvila, R.; Linnett, S.; et al. Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature 2018, 555, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.K.; Feil, R. Epigenetic transitions in germ cell development and meiosis. Dev. Cell 2010, 19, 675–686. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sakurai, T.; Imai, M.; Takahashi, N.; Fukuda, A.; Yayoi, O.; Sato, S.; Nakabayashi, K.; Hata, K.; Sotomaru, Y.; et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012, 8, e1002440. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019. [Google Scholar] [CrossRef]
- Goovaerts, T.; Steyaert, S.; Vandenbussche, C.A.; Galle, J.; Thas, O.; Van Criekinge, W.; De Meyer, T. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat. Commun. 2018, 9, 4120. [Google Scholar] [CrossRef]
- Hiura, H.; Obata, Y.; Komiyama, J.; Shirai, M.; Kono, T. Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells 2006, 11, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahurova, L.; Tomizawa, S.I.; Smallwood, S.A.; Stewart-Morgan, K.R.; Saadeh, H.; Kim, J.; Andrews, S.R.; Chen, T.; Kelsey, G. Transcription and chromatin determinants of de novo DNA methylation timing in oocytes. Epigenet. Chromatin 2017, 10, 25. [Google Scholar] [CrossRef]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.R.; Veselovska, L.; Kim, J.; Huang, J.; Saadeh, H.; Tomizawa, S.; Smallwood, S.A.; Chen, T.; Kelsey, G. Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev. 2015, 29, 2449–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannis, G.; Niederhuth, C.E.; Tuna, S.; Stathopoulou, A.; Viiri, K.; de Rooij, D.G.; Jenner, R.G.; Schmitz, R.J.; Ooi, S.K. The Dnmt3L ADD Domain Controls Cytosine Methylation Establishment during Spermatogenesis. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chotalia, M.; Smallwood, S.A.; Ruf, N.; Dawson, C.; Lucifero, D.; Frontera, M.; James, K.; Dean, W.; Kelsey, G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009, 23, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Engemann, S.; Lane, N.; Mayer, W.; Olek, A.; Fundele, R.; Dean, W.; Reik, W.; Walter, J. Active demethylation of the paternal genome in the mouse zygote. Curr. Biol. 2000, 10, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Jin, S.G.; Pfeifer, G.P.; Szabo, P.E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar] [CrossRef] [Green Version]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Zhang, Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 2011, 334, 194. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Shen, L.; Dai, Q.; He, C.; Zhang, Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011, 21, 1670–1676. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Liu, Y.J.; Nakashima, H.; Umehara, H.; Inoue, K.; Matoba, S.; Tachibana, M.; Ogura, A.; Shinkai, Y.; Nakano, T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 2012, 486, 415–419. [Google Scholar] [CrossRef]
- Cardoso, M.C.; Leonhardt, H. DNA methyltransferase is actively retained in the cytoplasm during early development. J. Cell Biol. 1999, 147, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Howell, C.Y.; Bestor, T.H.; Ding, F.; Latham, K.E.; Mertineit, C.; Trasler, J.M.; Chaillet, J.R. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 2001, 104, 829–838. [Google Scholar] [CrossRef]
- Hirasawa, R.; Chiba, H.; Kaneda, M.; Tajima, S.; Li, E.; Jaenisch, R.; Sasaki, H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008, 22, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef]
- Mackay, D.J.; Callaway, J.L.; Marks, S.M.; White, H.E.; Acerini, C.L.; Boonen, S.E.; Dayanikli, P.; Firth, H.V.; Goodship, J.A.; Haemers, A.P.; et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 2008, 40, 949–951. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; de Vries, W.; Ito, M.; Solter, D.; Ferguson-Smith, A.; Knowles, B.B. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012, 335, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Arai, Y.; Umehara, H.; Masuhara, M.; Kimura, T.; Taniguchi, H.; Sekimoto, T.; Ikawa, M.; Yoneda, Y.; Okabe, M.; et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol. 2007, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Auclair, G.; Guibert, S.; Bender, A.; Weber, M. Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse. Genome Biol. 2014, 15, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgel, J.; Guibert, S.; Li, Y.; Chiba, H.; Schubeler, D.; Sasaki, H.; Forne, T.; Weber, M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010, 42, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Chen, T. Mechanistic and functional links between histone methylation and DNA methylation. Prog. Mol. Biol. Transl. Sci. 2011, 101, 335–348. [Google Scholar] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Guenatri, M.; Duffie, R.; Iranzo, J.; Fauque, P.; Bourc’his, D. Plasticity in Dnmt3L-dependent and -independent modes of de novo methylation in the developing mouse embryo. Development 2013, 140, 562–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. https://doi.org/10.3390/genes10040257
Zeng Y, Chen T. DNA Methylation Reprogramming during Mammalian Development. Genes. 2019; 10(4):257. https://doi.org/10.3390/genes10040257
Chicago/Turabian StyleZeng, Yang, and Taiping Chen. 2019. "DNA Methylation Reprogramming during Mammalian Development" Genes 10, no. 4: 257. https://doi.org/10.3390/genes10040257
APA StyleZeng, Y., & Chen, T. (2019). DNA Methylation Reprogramming during Mammalian Development. Genes, 10(4), 257. https://doi.org/10.3390/genes10040257