Effect of Disease-Associated Germline Mutations on Structure Function Relationship of DNA Methyltransferases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
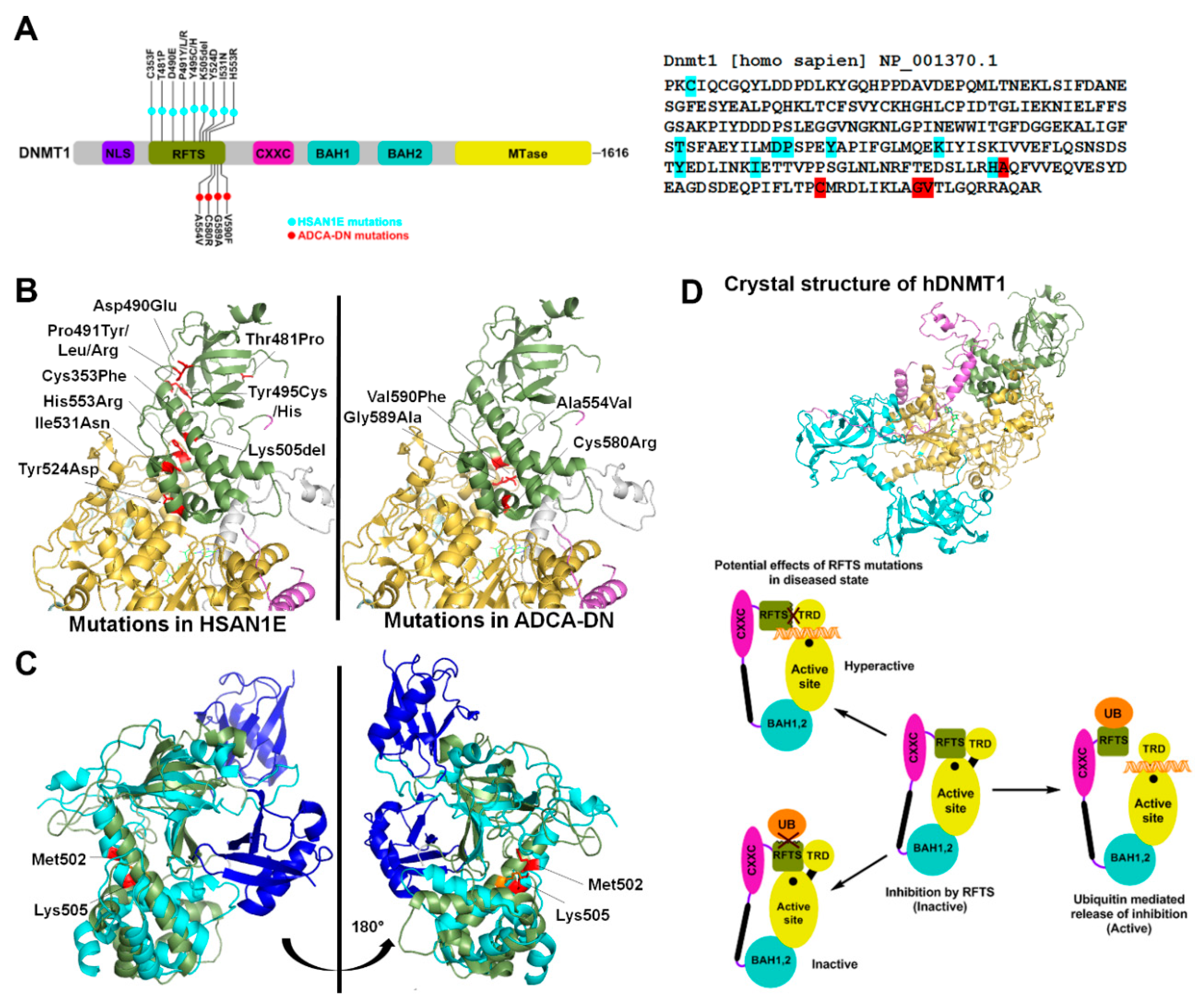
2. Structural and Functional Alterations of DNMT1 by Disease Associated Mutations
2.1. HSAN1E
2.2. ADCA-DN
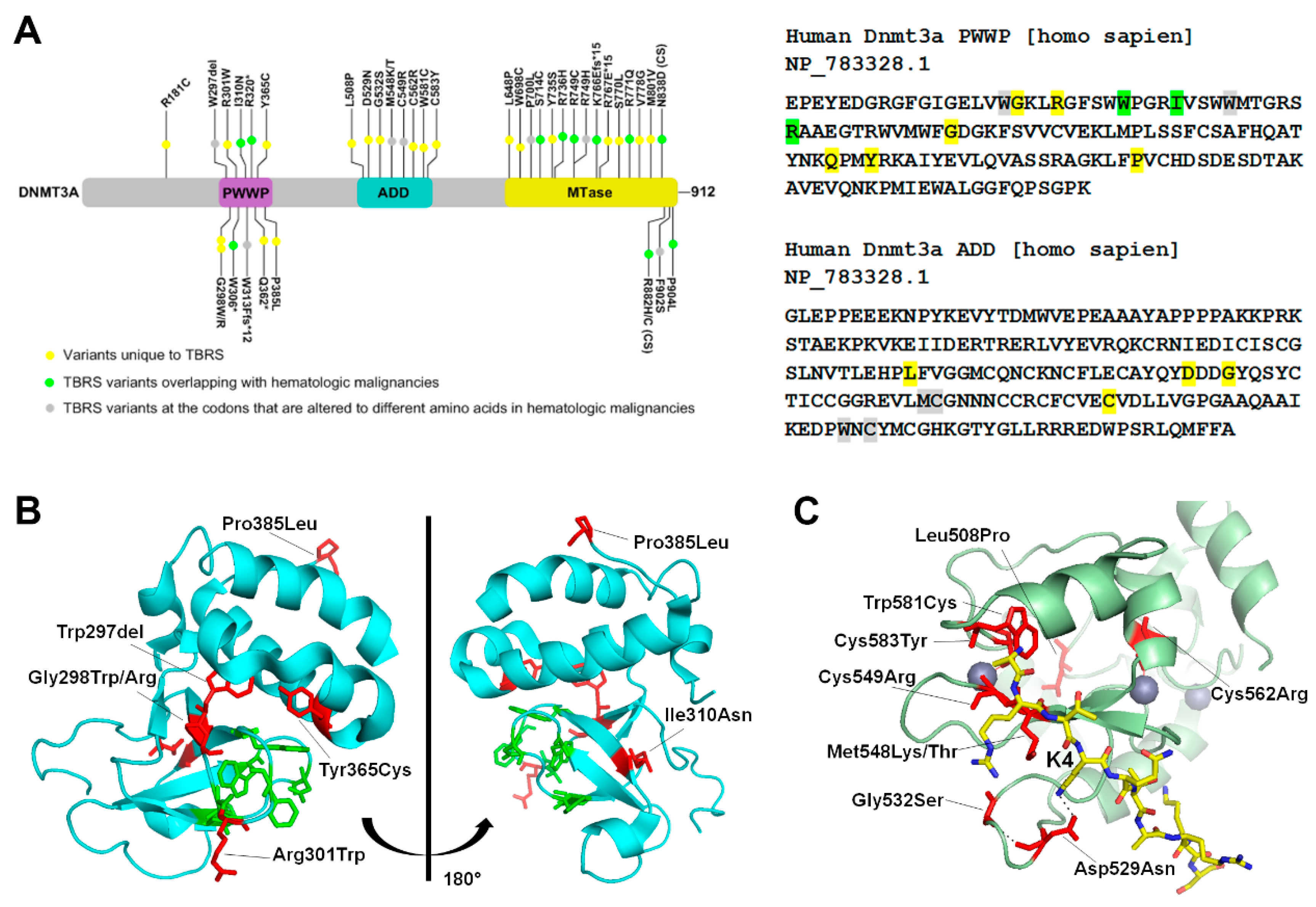
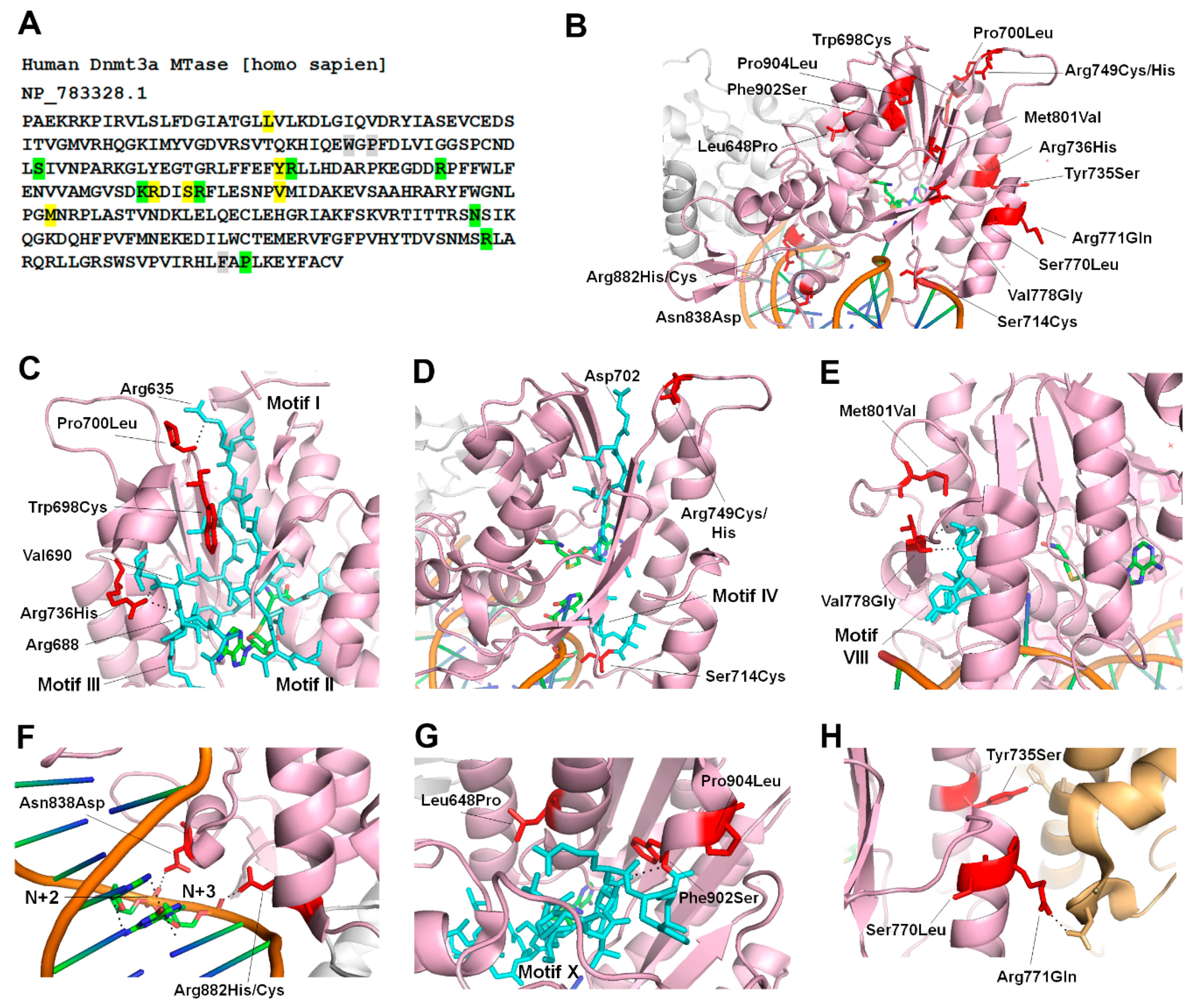
3. Structural and Functional Alterations of DNMT3 by Disease Associated Mutations
3.1. Tatton-Brown-Rahman Syndrome
3.2. Hereditary Tumors and Microcephalic Dwarfism (MD)
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H.; Boulard, M. DNA methylation and DNA methyltransferases. Epigenetics Chromatin 2017, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Microbiol. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Microbiol. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, X.; Zhou, Y.; Lee, M.; Guo, L.; Han, W.; Mo, W.; Cao, W.-M.; Sun, D.; Xie, R.; et al. Decoding the dynamic DNA methylation and hydroxymethylation landscapes in endodermal lineage intermediates during pancreatic differentiation of hESC. Nucleic Acids 2018, 46, 2883–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahé, E.A.; Madigou, T.; Sérandour, A.A.; Bizot, M.; Avner, S.; Chalmel, F.; Palierne, G.; Métivier, R.; Salbert, G. Cytosine modifications modulate the chromatin architecture of transcriptional enhancers. Genome Res. 2017, 27, 947–958. [Google Scholar] [CrossRef]
- Hackett, J.A.; Dietmann, S.; Murakami, K.; Down, T.A.; Leitch, H.G.; Surani, M.A. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Rep. 2013, 1, 518–531. [Google Scholar] [CrossRef]
- Cheng, X. Structure and function of DNA methyltransferases. Annu. Biophys. Biomol. Struct. 1995, 24, 293–318. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Mammalian DNA methyltransferases: New discoveries and open questions. Biochem. Soc. Trans. 2018, 46, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Li, E. Establishment and maintenance of DNA methylation patterns in mammals. In DNA Methylation: Basic Mechanisms; Springer: Berlin/Heidelberg, Germany, 2006; pp. 179–201. [Google Scholar]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Boil. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jeltsch, A. Enzymology of mammalian DNA methyltransferases. Purine Pyrimidine Metab. Man V 2016, 945, 87–122. [Google Scholar]
- Tajima, S.; Suetake, I.; Takeshita, K.; Nakagawa, A.; Kimura, H. Domain structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA methyltransferases. Adv. Exp. Med. Boil. 2016, 945, 63–86. [Google Scholar]
- Jones, P.A.; Issa, J.-P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Microbiol. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Velasco, G.; Francastel, C. Genetics meets DNA methylation in rare diseases. Clin. Genet. 2019, 95, 210–220. [Google Scholar] [CrossRef]
- Sandoval, J.E.; Huang, Y.H.; Muise, A.; Goodell, M.A.; Reich, N.O. Mutations in the DNMT3A DNA methyltransferase in AML patients cause both loss and gain of function and differential regulation by protein partners. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Norvil, A.B.; Petell, C.J.; Alabdi, L.; Wu, L.; Rossie, S.; Gowher, H. Dnmt3b methylates DNA by a noncooperative mechanism, and its activity is unaffected by manipulations at the predicted dimer interface. Biochemistry 2016, 57, 4312–4324. [Google Scholar] [CrossRef]
- Emperle, M.; Dukatz, M.; Kunert, S.; Holzer, K.; Rajavelu, A.; Jurkowska, R.Z.; Jeltsch, A. The DNMT3A R882H mutation does not cause dominant negative effects in purified mixed DNMT3A/R882H complexes. Sci. Rep. 2018, 8, 13242. [Google Scholar] [CrossRef] [PubMed]
- Emperle, M.; Rajavelu, A.; Kunert, S.; Arimondo, P.B.; Reinhardt, R.; Jurkowska, R.Z.; Jeltsch, A. The DNMT3A R882H mutant displays altered flanking sequence preferences. Nucleic Acids 2018, 46, 3130–3139. [Google Scholar] [CrossRef] [PubMed]
- Holz-Schietinger, C.; Matje, D.M.; Reich, N.O. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Boil. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Rechkoblit, O.; Bestor, T.H.; Patel, D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011, 331, 1036–1040. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Estève, P.-O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef]
- Svedruzic, Z.M. Dnmt1 structure and function. Prog. Mol. Biol. Transl. Sci. 2011, 101, 221–254. [Google Scholar] [PubMed]
- Baets, J.; Duan, X.; Wu, Y.; Smith, G.; Mademan, I.; Khoury, J.; Botuyan, M.-V.; Mer, G.; Hojo, K.; DeLeon, J.; et al. Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain 2015, 138, 845–861. [Google Scholar] [CrossRef]
- Winkelmann, J.; Lin, L.; Schormair, B.; Kornum, B.R.; Faraco, J.; Plazzi, G.; Melberg, A.; Cornelio, F.; Urban, A.E.; Pizza, F.; et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 2012, 21, 2205–2210. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.J.; Botuyan, M.-V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Wu, Y.; Ordog, T.; Baheti, S.; Nie, J.; Duan, X.; Hojo, K.; Kocher, J.-P.; Dyck, P.J.; Klein, C.J. Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics 2014, 9, 1184–1193. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.-K.; Mei, S.-C.; Brenner, C. RFTS-deleted DNMT1 enhances tumorigenicity with focal hypermethylation and global hypomethylation. Cell Cycle 2014, 13, 3222–3231. [Google Scholar] [CrossRef] [Green Version]
- Smets, M.; Link, S.; Wolf, P.; Schneider, K.; Solis, V.; Ryan, J.; Meilinger, D.; Qin, W.; Leonhardt, H. DNMT1 mutations found in HSANIE patients affect interaction with UHRF1 and neuronal differentiation. Hum. Mol. Genet. 2017, 26, 1522–1534. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.J.; Bird, T.; Ertekin-Taner, N.; Lincoln, S.; Hjorth, R.; Wu, Y.; Kwok, J.; Mer, G.; Dyck, P.J.; Nicholson, G.A. DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 2013, 80, 824–828. [Google Scholar] [CrossRef]
- Moghadam, K.K.; Pizza, F.; La Morgia, C.; Franceschini, C.; Tonon, C.; Lodi, R.; Barboni, P.; Seri, M.; Ferrari, S.; Liguori, R.; et al. Narcolepsy is a common phenotype in HSAN IE and ADCA-DN. Brain 2014, 137, 1643–1655. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Higuchi, Y.; Nagado, T.; Nozuma, S.; Nakamura, T.; Matsuura, E.; Hashiguchi, A.; Sakiyama, Y.; Yoshimura, A.; Takashima, H. Novel mutation in the replication focus targeting sequence domain of DNMT1 causes hereditary sensory and autonomic neuropathy IE. J. Peripher. Nerv. 2013, 18, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-M.; Liu, S.; Lin, K.; Luo, Y.; Perry, J.J.; Wang, Y.; Song, J. Crystal structure of human DNA methyltransferase 1. J. Mol. Boil. 2015, 427, 2520–2531. [Google Scholar] [CrossRef]
- Li, T.; Wang, L.; Du, Y.; Xie, S.; Yang, X.; Lian, F.; Zhou, Z.; Qian, C. Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids 2018, 46, 3218–3231. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Barsottini, O.G.P.; Lin, L.; Melberg, A.; Oliveira, A.S.B.; Mignot, E. A Novel de novo exon 21 DNMT1 mutation causes cerebellar ataxia, deafness, and narcolepsy in a Brazilian patient. Sleep 2013, 36, 1257–1259. [Google Scholar] [CrossRef]
- Kernohan, K.D.; Care4Rare Canada Consortium; Schenkel, L.C.; Huang, L.; Smith, A.; Pare, G.; Ainsworth, P.; Boycott, K.M.; Warman-Chardon, J.; Sadikovic, B. Identification of a methylation profile for DNMT1-associated autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Clin. Epigenetics 2016, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef]
- Yokoyama, A.S.; Rutledge, J.C.; Medici, V. DNA methylation alterations in Alzheimer’s disease. Environ. Epigenetics 2017, 3, 008. [Google Scholar] [CrossRef]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates α-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein sequesters Dnmt1 from the nucleus a novel mechanism for epigenetic alterations in lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Wüllner, U.; Kaut, O.; deBoni, L.; Piston, D.; Schmitt, I. DNA methylation in Parkinson’s disease. J. Neurochem. 2016, 139, 108–120. [Google Scholar] [CrossRef]
- Weissman, J.; Naidu, S.; Bjornsson, H.T. Abnormalities of the DNA methylation mark and its machinery: An emerging cause of neurologic dysfunction. Semin. Neurol. 2014, 34, 249–257. [Google Scholar] [CrossRef]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Forbes, S.; Bhamra, G.; Bamford, S.; Dawson, E.; Kok, C.; Clements, J.; Menzies, A.; Teague, J.; Futreal, P.; Stratton, M. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr. Protoc. Hum. Genet. 2008, 57, Unit 10.11. [Google Scholar]
- Cheng, X.; Blumenthal, R.M. Mammalian DNA methyltransferases: A structural perspective. Structure 2008, 16, 341–350. [Google Scholar] [CrossRef]
- Chen, T.; Tsujimoto, N.; Li, E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Boil. 2004, 24, 9048–9058. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 Lysine 36 trimethylation and guides DNA methylation. J. Boil. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef]
- Wu, H.; Zeng, H.; Lam, R.; Tempel, W.; Amaya, M.F.; Xu, C.; Dombrovski, L.; Qiu, W.; Wang, Y.; Min, J. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 2011, 6, e18919. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nat. Cell Boil. 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wang, L.; Li, J.; Ding, Z.; Xiao, J.; Yin, X.; He, S.; Shi, P.; Dong, L.; Li, G.; et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 2015, 517, 640. [Google Scholar] [CrossRef] [PubMed]
- Petell, C.J.; Alabdi, L.; He, M.; Miguel, P.S.; Rose, R.; Gowher, H. An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids 2016, 44, 7605–7617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowher, H.; Loutchanwoot, P.; Vorobjeva, O.; Handa, V.; Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J. Mol. Boil. 2006, 357, 928–941. [Google Scholar] [CrossRef]
- Rajavelu, A.; Jurkowska, R.Z.; Fritz, J.; Jeltsch, A. Function and disruption of DNA Methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids 2011, 40, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nat. Cell Boil. 2007, 449, 248–251. [Google Scholar] [CrossRef] [Green Version]
- Emperle, M.; Rajavelu, A.; Reinhardt, R.; Jurkowska, R.Z.; Jeltsch, A. Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Boil. Chem. 2014, 289, 29602–29613. [Google Scholar] [CrossRef]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.R.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Acad. Sci. 1999, 96, 14412–14417. [Google Scholar] [CrossRef]
- Xu, G.-L.; Bestor, T.H.; Bourc’His, D.; Hsieh, C.-L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nat. Cell Boil. 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Ehrlich, M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin. Immunol. 2003, 109, 17–28. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Childhood Overgrowth Consortium; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Duarte, S.D.V.; Zachariou, A.; Hanks, S.; O’Brien, E.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Zachariou, A.; Loveday, C.; Renwick, A.; Mahamdallie, S.; Aksglaede, L.; Baralle, D.; Barge-Schaapveld, D.; Blyth, M.; Bouma, M.; et al. The Tatton-Brown-Rahman syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open 2018, 3, 46. [Google Scholar] [CrossRef]
- Okamoto, N.; Toribe, Y.; Shimojima, K.; Yamamoto, T. Tatton-Brown-Rahman syndrome due to 2p23 microdeletion. Am. J. Med. Genet. Genet. A 2016, 170, 1339–1342. [Google Scholar] [CrossRef]
- Hollink, I.H.; van den Ouweland, A.M.; Beverloo, H.B.; Arentsen-Peters, S.T.; Zwaan, C.M.; Wagner, A. Acute myeloid leukaemia in a case with Tatton-Brown-Rahman syndrome: The peculiar DNMT3A R882 mutation. J. Med. Genet. 2017, 54, 805–808. [Google Scholar] [CrossRef]
- Kosaki, R.; Terashima, H.; Kubota, M.; Kosaki, K. Acute myeloid leukemia-associated DNMT3A p. Arg882His mutation in a patient with Tatton-Brown–Rahman overgrowth syndrome as a constitutional mutation. Am. J. Med. Genet. Part A 2017, 173, 250–253. [Google Scholar] [CrossRef]
- Tlemsani, C.; Luscan, A.; Leulliot, N.; Bieth, E.; Afenjar, A.; Baujat, G.; Doco-Fenzy, M.; Goldenberg, A.; Lacombe, D.; Lambert, L.; et al. SETD2 and DNMT3A screen in the Sotos-like syndrome French cohort. J. Med. Genet. 2016, 53, 743–751. [Google Scholar] [CrossRef]
- Xin, B.; Marino, T.C.; Szekely, J.; Leblanc, J.; Cechner, K.; Sency, V.; Wensel, C.; Barabas, M.; Therriault, V.; Wang, H. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin. Genet. 2017, 91, 623–628. [Google Scholar] [CrossRef]
- Shen, W.; Heeley, J.M.; Carlston, C.M.; Acuna-Hidalgo, R.; Nillesen, W.M.; Dent, K.M.; Douglas, G.V.; Levine, K.L.; Bayrak-Toydemir, P.; Marcelis, C.L.; et al. The spectrum of DNMT3A variants in Tatton-Brown-Rahman syndrome overlaps with that in hematologic malignancies. Am. J. Med. Genet. Genet. A 2017, 173, 3022–3028. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, A.R.; Maroofian, R.; Salter, C.G.; Chioza, B.A.; Cross, H.E.; Patton, M.A.; Temple, I.K.; Mackay, D.; Rezwan, F.I.; Aksglaede, L.; et al. Growth disrupting mutations in epigenetic regulatory molecules are associated with abnormalities of epigenetic aging. bioRxiv 2018, 477356. [Google Scholar] [CrossRef]
- Zhang, Z.-M.; Lü, R.; Wang, P.; Yu, Y.; Chen, D.-L.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nat. Cell Boil. 2018, 554, 387–391. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A Mutations in Acute Myeloid Leukemia. New Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Zhao, H.; Hardikar, S.; Singh, A.K.; Goodell, M.A.; Chen, T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 2013, 122, 4086–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacha, L.; Currás-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Mendez, I.; Roldan-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018, 20, 1644–1651. [Google Scholar] [CrossRef]
- Averbuch, S.D.; Steakley, C.S.; Young, R.C.; Gelmann, E.P.; Goldstein, D.S.; Stull, R.; Keiser, H.R. Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann. Intern. Med. 1988, 109, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Heyn, P.; Logan, C.V.; Fluteau, A.; Challis, R.C.; Auchynnikava, T.; Martin, C.A.; Marsh, J.A.; Taglini, F.; Kilanowski, F.; Parry, D.A.; et al. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat. Genet. 2019, 51, 96–105. [Google Scholar] [CrossRef]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multi-lineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Boil. 2018, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17. [Google Scholar] [CrossRef]
- Long, H.K.; Sims, D.; Heger, A.; Blackledge, N.P.; Kutter, C.; Wright, M.L.; Grützner, F.; Odom, D.T.; Patient, R.; Ponting, C.P.; et al. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. eLife 2013, 2, 00348. [Google Scholar] [CrossRef]
- Wu, H.; Coskun, V.; Tao, J.; Xie, W.; Ge, W.; Yoshikawa, K.; Li, E.; Zhang, Y.; Sun, Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 2010, 329, 444–448. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; VasanthaKumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Shirohzu, H.; Kubota, T.; Kumazawa, A.; Sado, T.; Chijiwa, T.; Inagaki, K.; Suetake, I.; Tajima, S.; Wakui, K.; Miki, Y.; et al. Three novelDNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 2002, 112, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.-Z.; Gowher, H.; Jeltsch, A.; Pu, M.-T.; Wu, H.-P.; Ding, J.-P.; Xu, G.-L. Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J. Boil. Chem. 2004, 279, 25447–25454. [Google Scholar] [CrossRef]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nat. Cell Boil. 2015, 520, 243–247. [Google Scholar] [CrossRef]
- Duymich, C.E.; Charlet, J.; Yang, X.; Jones, P.A.; Liang, G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat. Commun. 2016, 7, 11453. [Google Scholar] [CrossRef] [Green Version]
- Van Emburgh, B.O.; Robertson, K.D. Modulation of Dnmt3b function in vitro by interactions with Dnmt3L, Dnmt3a and Dnmt3b splice variants. Nucleic Acids 2011, 39, 4984–5002. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Tao, Q.; Peng, J.; Soo, H.M.; Wu, W.; Ying, J.; Fields, C.R.; Delmas, A.L.; Liu, X.; Qiu, J.; et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008, 17, 690–709. [Google Scholar] [CrossRef]
- Maresca, A.; Zaffagnini, M.; Caporali, L.; Carelli, V.; Zanna, C. DNA methyltransferase 1 mutations and mitochondrial pathology: Is mtDNA methylated? Front. Genet. 2015, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norvil, A.B.; Saha, D.; Dar, M.S.; Gowher, H. Effect of Disease-Associated Germline Mutations on Structure Function Relationship of DNA Methyltransferases. Genes 2019, 10, 369. https://doi.org/10.3390/genes10050369
Norvil AB, Saha D, Dar MS, Gowher H. Effect of Disease-Associated Germline Mutations on Structure Function Relationship of DNA Methyltransferases. Genes. 2019; 10(5):369. https://doi.org/10.3390/genes10050369
Chicago/Turabian StyleNorvil, Allison B., Debapriya Saha, Mohd Saleem Dar, and Humaira Gowher. 2019. "Effect of Disease-Associated Germline Mutations on Structure Function Relationship of DNA Methyltransferases" Genes 10, no. 5: 369. https://doi.org/10.3390/genes10050369
APA StyleNorvil, A. B., Saha, D., Dar, M. S., & Gowher, H. (2019). Effect of Disease-Associated Germline Mutations on Structure Function Relationship of DNA Methyltransferases. Genes, 10(5), 369. https://doi.org/10.3390/genes10050369