ATM Serine/Threonine Kinase and its Role in Pancreatic Risk

Abstract
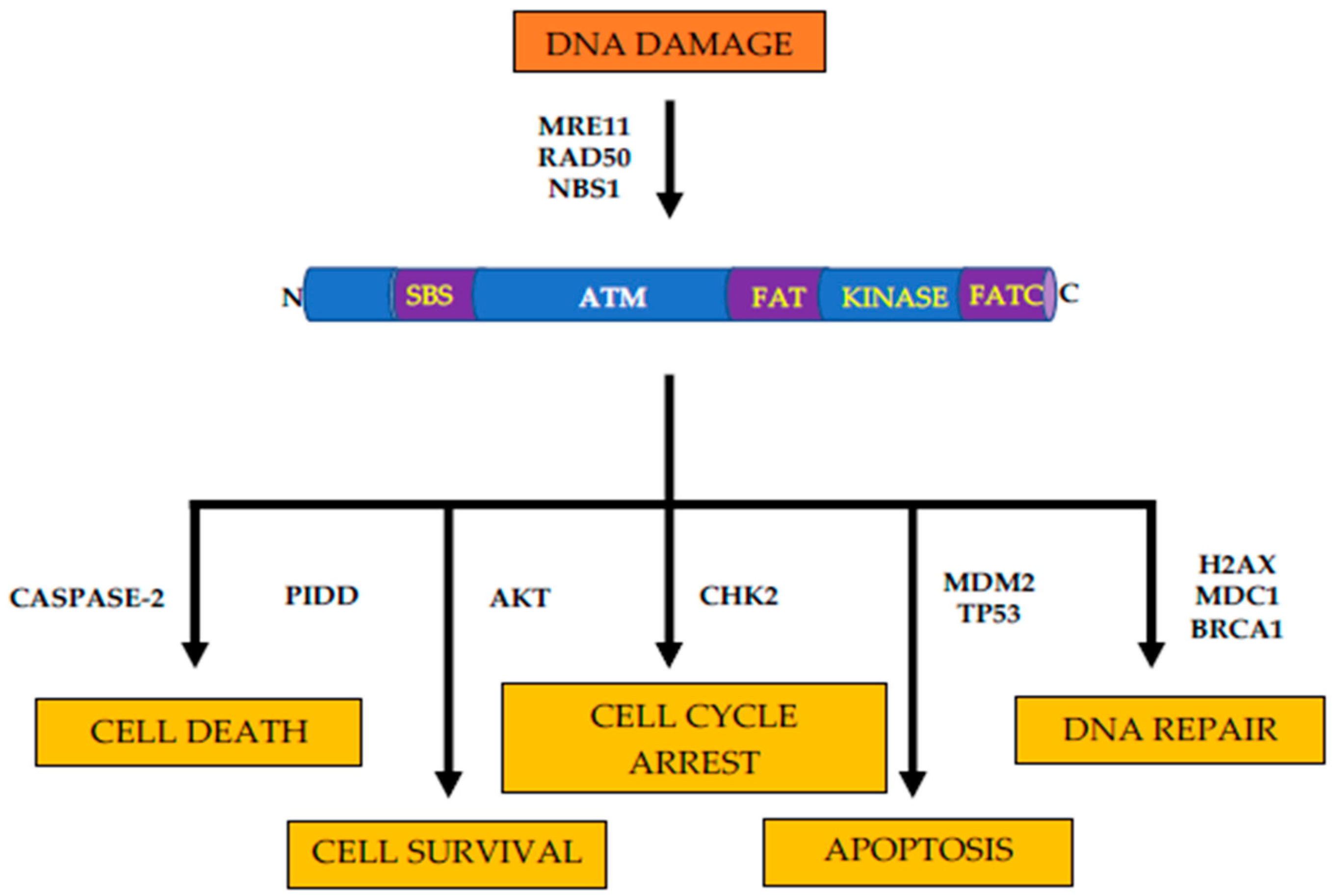
:1. Introduction
2. Pathogenic Germline ATM Variants in Patients with Familial Pancreatic Cancer
3. Pathogenic Germline ATM Variants in Patients without a Family History of Pancreatic Cancer
4. Pathogenic Germline ATM Variants in Patients with PanIN and IPMN
5. Somatic ATM Alterations in PDAC
6. Diagnostic and Therapeutic Implications
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients with Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [Green Version]
- Brune, K.A.; Lau, B.; Palmisano, E.; Canto, M.; Goggins, M.G.; Hruban, R.H.; Klein, A.P. Importance of age of onset in pancreatic cancer kindreds. J. Natl. Cancer Inst. 2010, 102, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Lucas, A.L.; Chang, M.M.; Lipsyc, M.D.; Frucht, H. The prevention and genetics of pancreatic cancer: A programmatic approach. Methods Mol. Biol. 2013, 980, 205–214. [Google Scholar]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.P. Identifying people at a high risk of developing pancreatic cancer. Nat. Rev. Cancer 2013, 13, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Lavin, M.F.; Scott, S.; Gueven, N.; Kozlov, S.; Peng, C.; Chen, P. Functional consequences of sequence alterations in the ATM gene. DNA Repair (Amst.) 2004, 3, 1197–1205. [Google Scholar] [CrossRef]
- Pizarro, J.G.; Folch, J.; de la Torre, A.V.; Junyent, F.; Verdaguer, E.; Jordan, J.; Pallas, M.; Camins, A. ATM is involved in cell-cycle control through the regulation of retinoblastoma protein phosphorylation. J. Cell. Biochem. 2010, 110, 210–218. [Google Scholar] [CrossRef]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227. [Google Scholar] [CrossRef]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef] [Green Version]
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583. [Google Scholar]
- Taylor, A.M.; Byrd, P.J. Molecular pathology of ataxia telangiectasia. J. Clin. Pathol. 2005, 58, 1009–1015. [Google Scholar] [CrossRef]
- Mavrou, A.; Tsangaris, G.T.; Roma, E.; Kolialexi, A. The ATM gene and ataxia telangiectasia. Anticancer Res. 2008, 28, 401–405. [Google Scholar]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Takai, E.; Yachida, S.; Shimizu, K.; Furuse, J.; Kubo, E.; Ohmoto, A.; Suzuki, M.; Hruban, R.H.; Okusaka, T.; Morizane, C.; et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget 2016, 7, 74227–74235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Hu, C.; LaDuca, H.; Shimelis, H.; Polley, E.C.; Lilyquist, J.; Hart, S.N.; Na, J.; Thomas, A.; Lee, K.Y.; Davis, B.T.; et al. Multigene Hereditary Cancer Panels Reveal High-Risk Pancreatic Cancer Susceptibility Genes. JCO Precis. Oncol. 2018, 2, 1–28. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.R.; Rotunno, M.; Xiao, Y.; Ingvar, C.; Helgadottir, H.; Pastorino, L.; van Doorn, R.; Bennett, H.; Graham, C.; Sampson, J.N.; et al. Multiple rare variants in high-risk pancreatic cancer-related genes may increase risk for pancreatic cancer in a subset of patients with and without germline CDKN2A mutations. Hum. Genet. 2016, 135, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients with Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs. Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.; Borazanci, E.; Speare, V.; Dudley, B.; Karloski, E.; Peters, M.L.B.; Stobie, L.; Bahary, N.; Zeh, H.; Zureikat, A.; et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer 2018, 124, 3520–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.L.; Wong, C.; Cuggia, A.; Borgida, A.; Holter, S.; Hall, A.; Connor, A.A.; Bascuñana, C.; Asselah, J.; Bouganim, N.; et al. Reflex Testing for Germline BRCA1, BRCA2, PALB2, and ATM Mutations in Pancreatic Cancer: Mutation Prevalence and Clinical Outcomes from Two Canadian Research Registries. JCO Precis. Oncol. 2018, 2, 1–16. [Google Scholar] [CrossRef]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients with Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempero, M.A.; Malafa, M.P.; Chiorean, E.G.; Czito, B.; Scaife, C.; Narang, A.K.; Fountzilas, C.; Wolpin, B.M.; Al-Hawary, M.; Asbun, H.; et al. Pancreatic Adenocarcinoma, Version 1.2019. J. Natl. Compr. Cancer Netw. 2019, 17, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelker, D.; Donoghue, M.; Talukdar, S.; Bandlamudi, C.; Srinivasan, P.; Vivek, M.; Jezdic, S.; Hanson, H.; Snape, K.; Kulkarni, A.; et al. Germline-focussed analysis of tumour-only sequencing: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2019, 30, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.Y.; Yu, Y.D.; Kim, W.B.; Han, H.J.; Choi, S.B.; Kim, D.S.; Choi, S.Y.; Kim, J.Y.; Chang, H.; Kim, B.H. Clinical significance of pancreatic intraepithelial neoplasia in resectable pancreatic cancer on survivals. Ann. Surg. Treat. Res. 2018, 94, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.J.; Hart, S.N.; Lima, J.F.; Kipp, B.R.; Klebig, M.; Winters, J.L.; Szabo, C.; Zhang, L.; Eckloff, B.W.; Petersen, G.M.; et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013, 145, 1098–1109. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Guthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [Green Version]
- Laffan, T.A.; Horton, K.M.; Klein, A.P.; Berlanstein, B.; Siegelman, S.S.; Kawamoto, S.; Johnson, P.T.; Fishman, E.K.; Hruban, R.H. Prevalence of unsuspected pancreatic cysts on MDCT. AJR Am. J. Roentgenol. 2008, 191, 802–807. [Google Scholar] [CrossRef] [Green Version]
- Skaro, M.; Nanda, N.; Gauthier, C.; Felsenstein, M.; Jiang, Z.; Qiu, M.; Shindo, K.; Yu, J.; Hutchings, D.; Javed, A.A.; et al. Prevalence of Germline Mutations Associated with Cancer Risk in Patients with Intraductal Papillary Mucinous Neoplasms. Gastroenterology 2019, 156, 1905–1913. [Google Scholar] [CrossRef]
- Grant, R.C.; Al-Sukhni, W.; Borgida, A.E.; Holter, S.; Kanji, Z.S.; McPherson, T.; Whelan, E.; Serra, S.; Trinh, Q.M.; Peltekova, V.; et al. Exome sequencing identifies nonsegregating nonsense ATM and PALB2 variants in familial pancreatic cancer. Hum. Genom. 2013, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Sikdar, N.; Saha, G.; Dutta, A.; Ghosh, S.; Shrikhande, S.V.; Banerjee, S. Genetic Alterations of Periampullary and Pancreatic Ductal Adenocarcinoma: An Overview. Curr. Genom. 2018, 19, 444–463. [Google Scholar] [CrossRef]
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 2015, 7, 283ra53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A., Jr.; Papadopoulos, N.; et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013, 45, 12–17. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Valero III, V.; Saunders, T.J.; He, J.; Weiss, M.J.; Cameron, J.L.; Dholakia, A.; Wild, A.T.; Shin, E.J.; Khashab, M.A.; O’Broin-Lennon, A.M.; et al. Reliable Detection of Somatic Mutations in Fine Needle Aspirates of Pancreatic Cancer with Next-generation Sequencing: Implications for Surgical Management. Ann. Surg. 2016, 263, 153–161. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Saka, B.; Knight, S.; Borges, M.; Childs, E.; Klein, A.; Wolfgang, C.; Herman, J.; Adsay, V.N.; Hruban, R.H.; et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin. Cancer Res. 2014, 20, 1865–1872. [Google Scholar] [CrossRef] [Green Version]
- Drosos, Y.; Escobar, D.; Chiang, M.Y.; Roys, K.; Valentine, V.; Valentine, M.B.; Rehg, J.E.; Sahai, V.; Begley, L.A.; Ye, J.; et al. ATM-deficiency increases genomic instability and metastatic potential in a mouse model of pancreatic cancer. Sci. Rep. 2017, 7, 11144. [Google Scholar] [CrossRef]
- Jiao, Y.; Yonescu, R.; Offerhaus, G.J.; Klimstra, D.S.; Maitra, A.; Eshleman, J.R.; Herman, J.G.; Poh, W.; Pelosof, L.; Wolfgang, C.L.; et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J. Pathol. 2014, 232, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Corbo, V.; Ritelli, R.; Barbi, S.; Funel, N.; Campani, D.; Bardelli, A.; Scarpa, A. Mutational profiling of kinases in human tumours of pancreatic origin identifies candidate cancer genes in ductal and ampulla of vater carcinomas. PLoS ONE 2010, 5, e12653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2018. Available online: http://seer.cancer.gov/csr/1975_2015/ (accessed on 17 January 2020).
- Vasen, H.; Ibrahim, I.; Ponce, C.G.; Slater, E.P.; Matthai, E.; Carrato, A.; Earl, J.; Robbers, K.; van Mil, A.M.; Potjer, T.; et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies from Three European Expert Centers. J. Clin. Oncol. 2016, 34, 2010–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Blackford, A.L.; Tamura, K.; Ford, M.; McCormick, P.; Chuidian, M.; Almario, J.A.; Borges, M.; Lennon, A.M.; Shin, E.J.; et al. Deleterious Germline Mutations Are a Risk Factor for Neoplastic Progression Among High-Risk Individuals Undergoing Pancreatic Surveillance. J. Clin. Oncol. 2019, 37, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Renault, A.L.; Mebirouk, N.; Fuhrmann, L.; Bataillon, G.; Cavaciuti, E.; Le Gal, D.; Girard, E.; Popova, T.; La Rosa, P.; Beauvallet, J.; et al. Morphology and genomic hallmarks of breast tumours developed by ATM deleterious variant carriers. Breast Cancer Res. 2018, 20, 28. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, D.; Jiang, Z.; Skaro, M.; Weiss, M.J.; Wolfgang, C.L.; Makary, M.A.; He, J.; Cameron, J.L.; Zheng, L.; Klimstra, D.S.; et al. Histomorphology of pancreatic cancer in patients with inherited ATM serine/threonine kinase pathogenic variants. Mod. Pathol. 2019, 32, 1806–1813. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures with Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Ayars, M.; Eshleman, J.; Goggins, M. Susceptibility of ATM-deficient pancreatic cancer cells to radiation. Cell Cycle 2017, 16, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesha, V.A.; Parsels, L.A.; Parsels, J.D.; Zhao, L.; Zabludoff, S.D.; Simeone, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia 2012, 14, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Patients | Total Number of Patients | Number of Pathogenic Germline ATM Variants | Percentage (%) | Reference |
|---|---|---|---|---|
| FPC | 166 | 4 | 2.4 | [19] |
| 54 | 2 | 3.7 | [22] | |
| 638 | 21 | 3.4 | [24] | |
| 185 | 6 | 3.2 | [8] | |
| Nonfamilial PDAC | 854 | 10 | 1.2 | [3] |
| 149 | 3 | 2 | [30] | |
| 117 | 2 | 1.7 | [8] | |
| 350 | 4 | 1.3 | [32] | |
| Unselected PDAC | 290 | 3 | 1.0 | [21] |
| 96 | 4 | 4.2 | [23] | |
| 176 | 5 | 2.8 | [29] | |
| 3030 | 69 | 2.3 | [25] | |
| 1213 | 46 | 3.8 | [26] | |
| 298 | 10 | 3.3 | [31] | |
| 615 | 11 | 1.8 | [33] | |
| 3594 | 48 | 1.3 | [34] | |
| 289 | 4 | 1.4 | [27] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanda, N.; Roberts, N.J. ATM Serine/Threonine Kinase and its Role in Pancreatic Risk. Genes 2020, 11, 108. https://doi.org/10.3390/genes11010108
Nanda N, Roberts NJ. ATM Serine/Threonine Kinase and its Role in Pancreatic Risk. Genes. 2020; 11(1):108. https://doi.org/10.3390/genes11010108
Chicago/Turabian StyleNanda, Neha, and Nicholas J. Roberts. 2020. "ATM Serine/Threonine Kinase and its Role in Pancreatic Risk" Genes 11, no. 1: 108. https://doi.org/10.3390/genes11010108
APA StyleNanda, N., & Roberts, N. J. (2020). ATM Serine/Threonine Kinase and its Role in Pancreatic Risk. Genes, 11(1), 108. https://doi.org/10.3390/genes11010108