Population Genomics in Rhamdia quelen (Heptapteridae, Siluriformes) Reveals Deep Divergence and Adaptation in the Neotropical Region

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
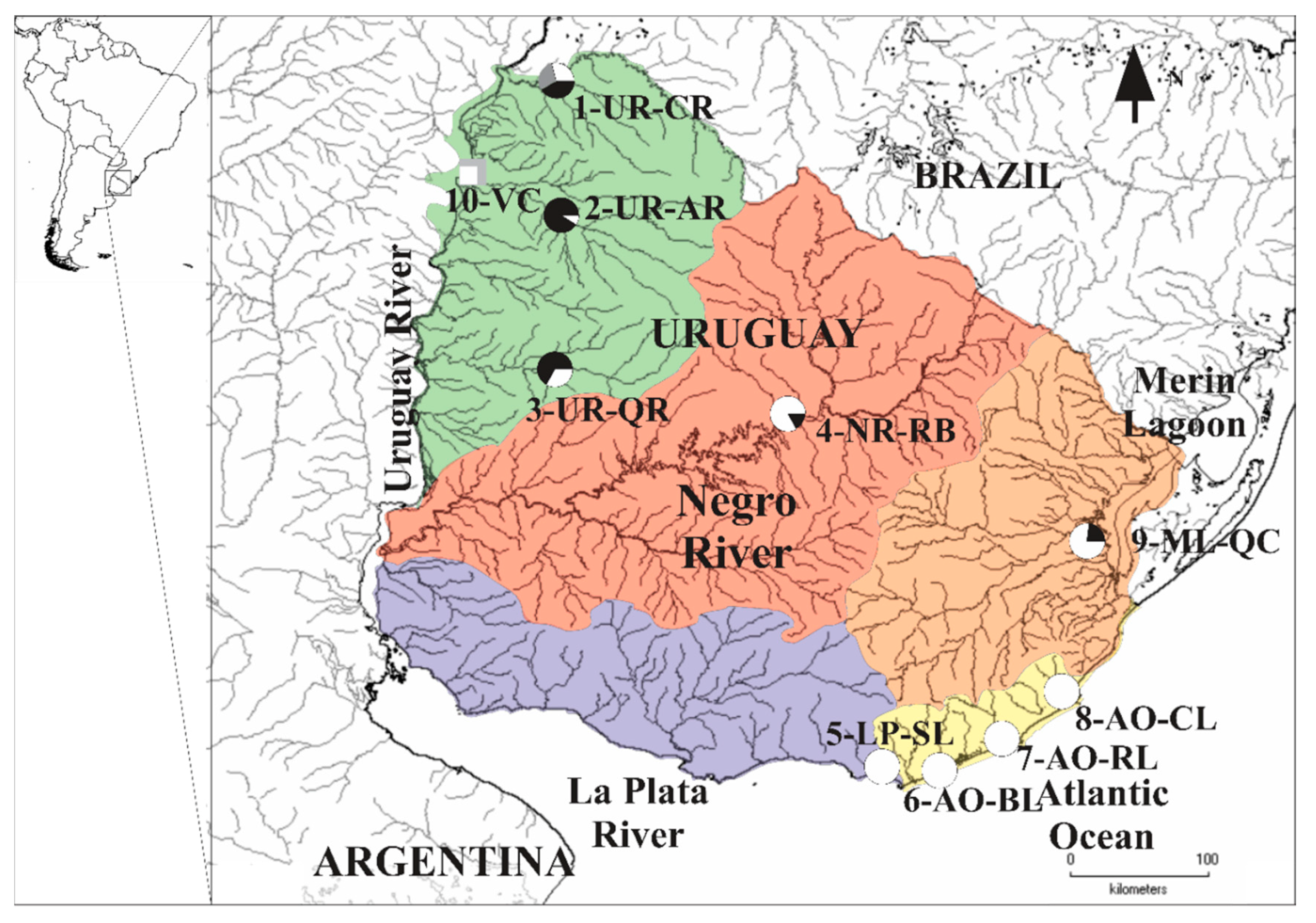
2.1. Sample Collection and DNA Extraction
2.2. Identification of Rhamdia quelen mtDNA Lineages Based on Partial Sequences of Cytb and COI
2.3. Library Construction and Sequencing
2.4. Data Processing and SNP Genotyping
2.5. Detection of Loci under Selection
2.6. Genetic Diversity and Population Structure of R. quelen in LP-PM-AO
2.7. Genetic Differentiation by Environmental Factors
2.8. Evolutionary History of Populations
3. Results
3.1. MtDNA Lineages Identification Based on cytb and COI
3.2. Genome Data Processing and SNP Genotyping
3.3. Detection of Putative Loci under Selection
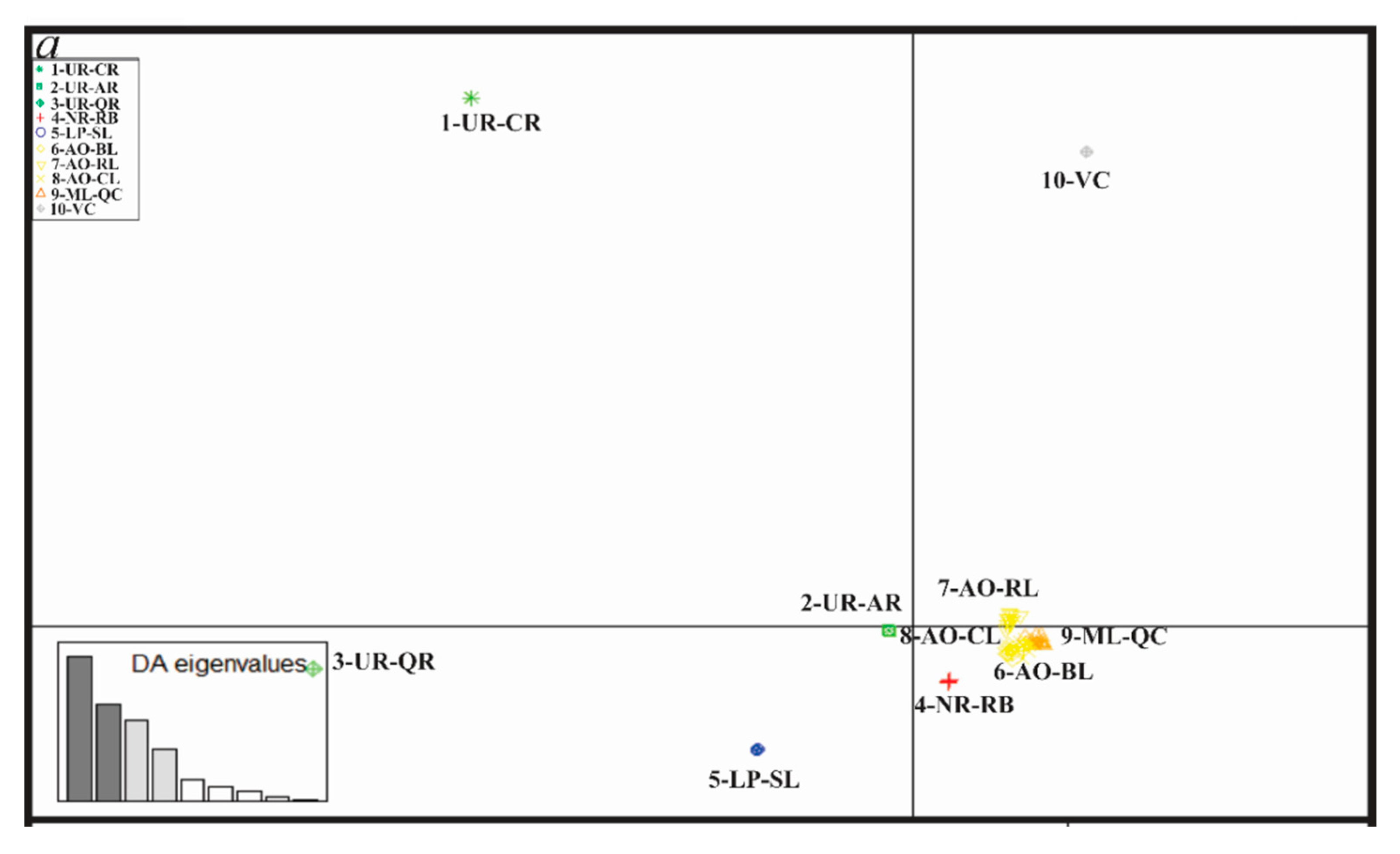
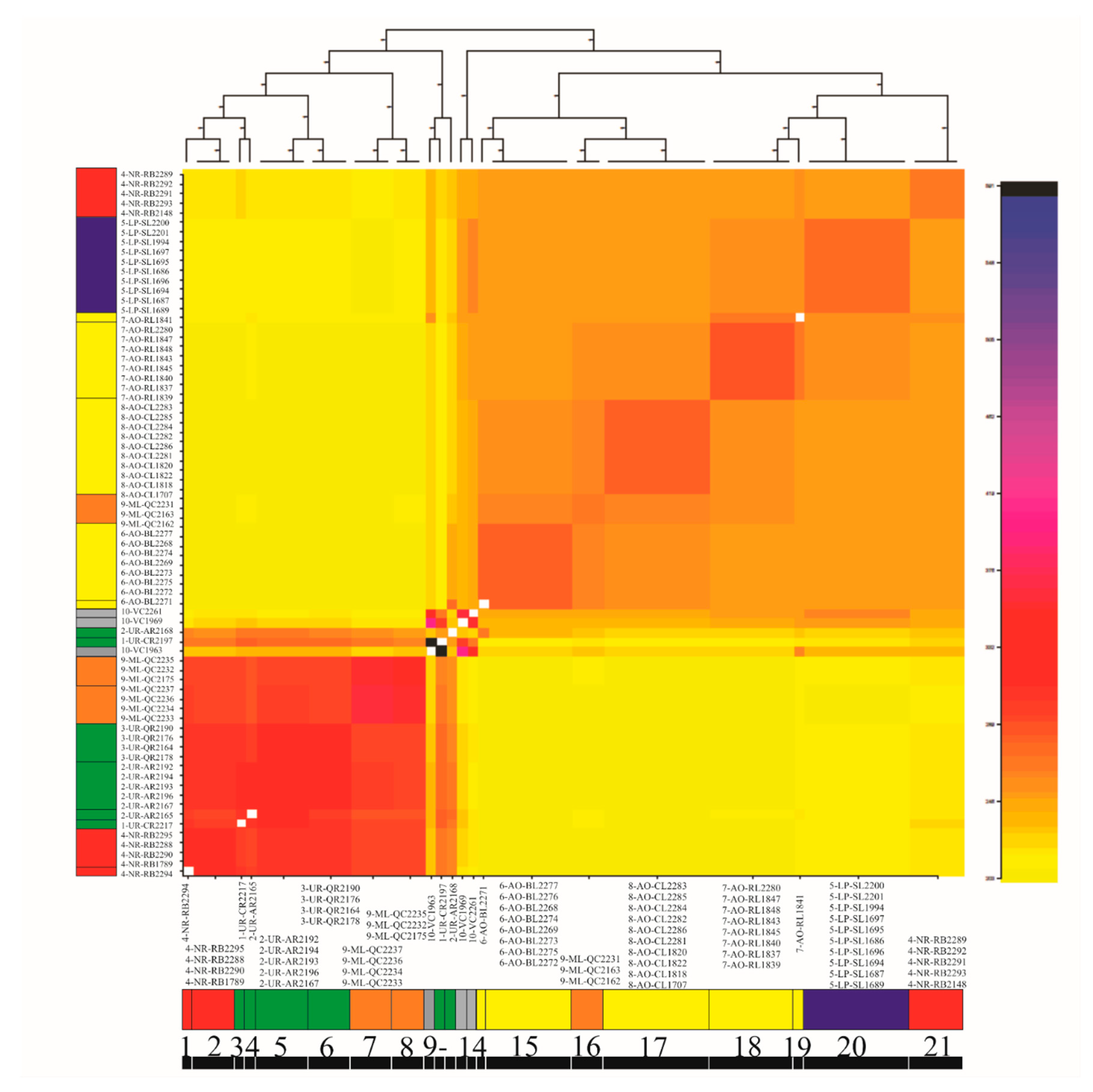
3.4. Genetic Diversity and Structure of R. quelen in LP-PM-AO
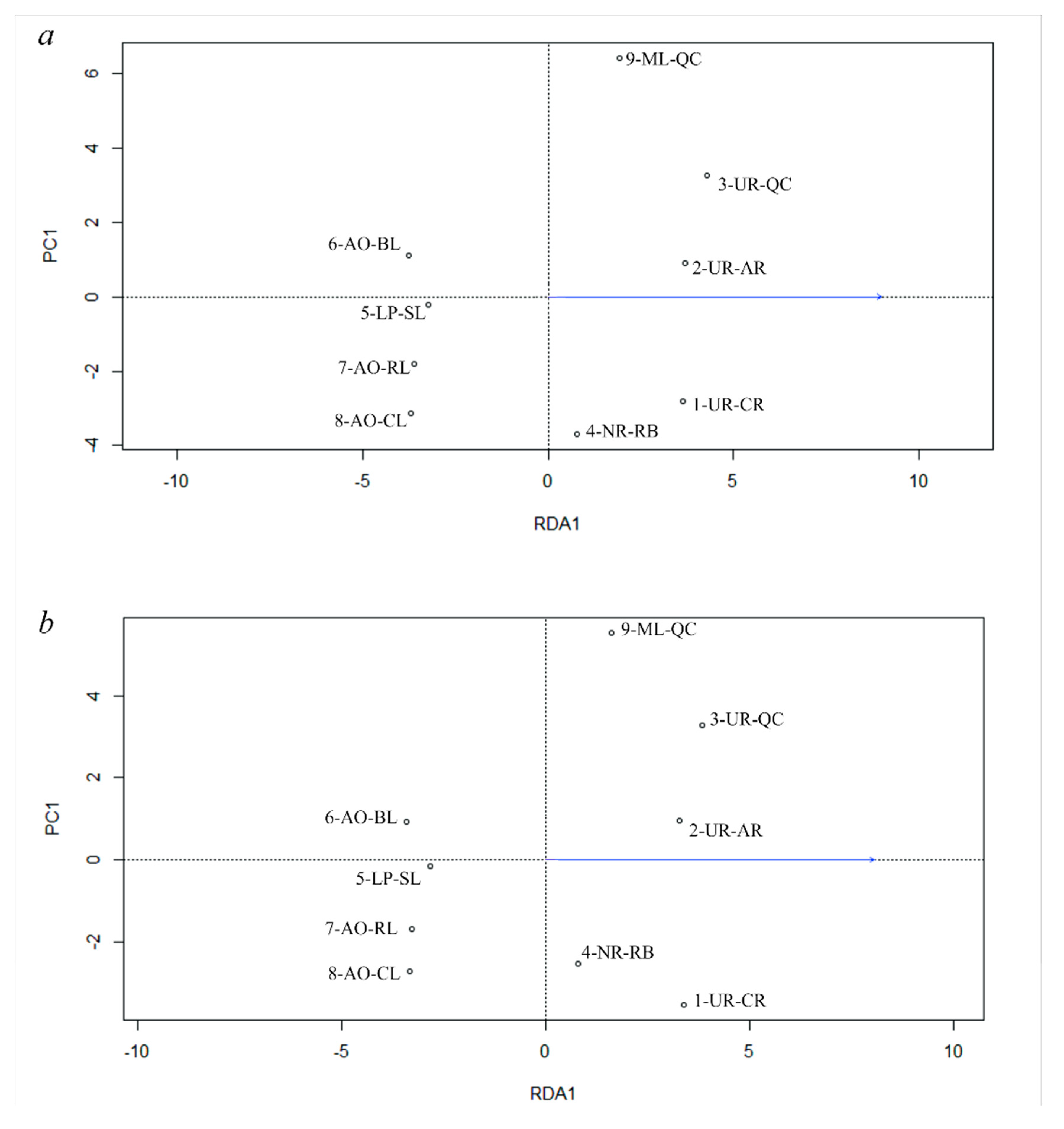
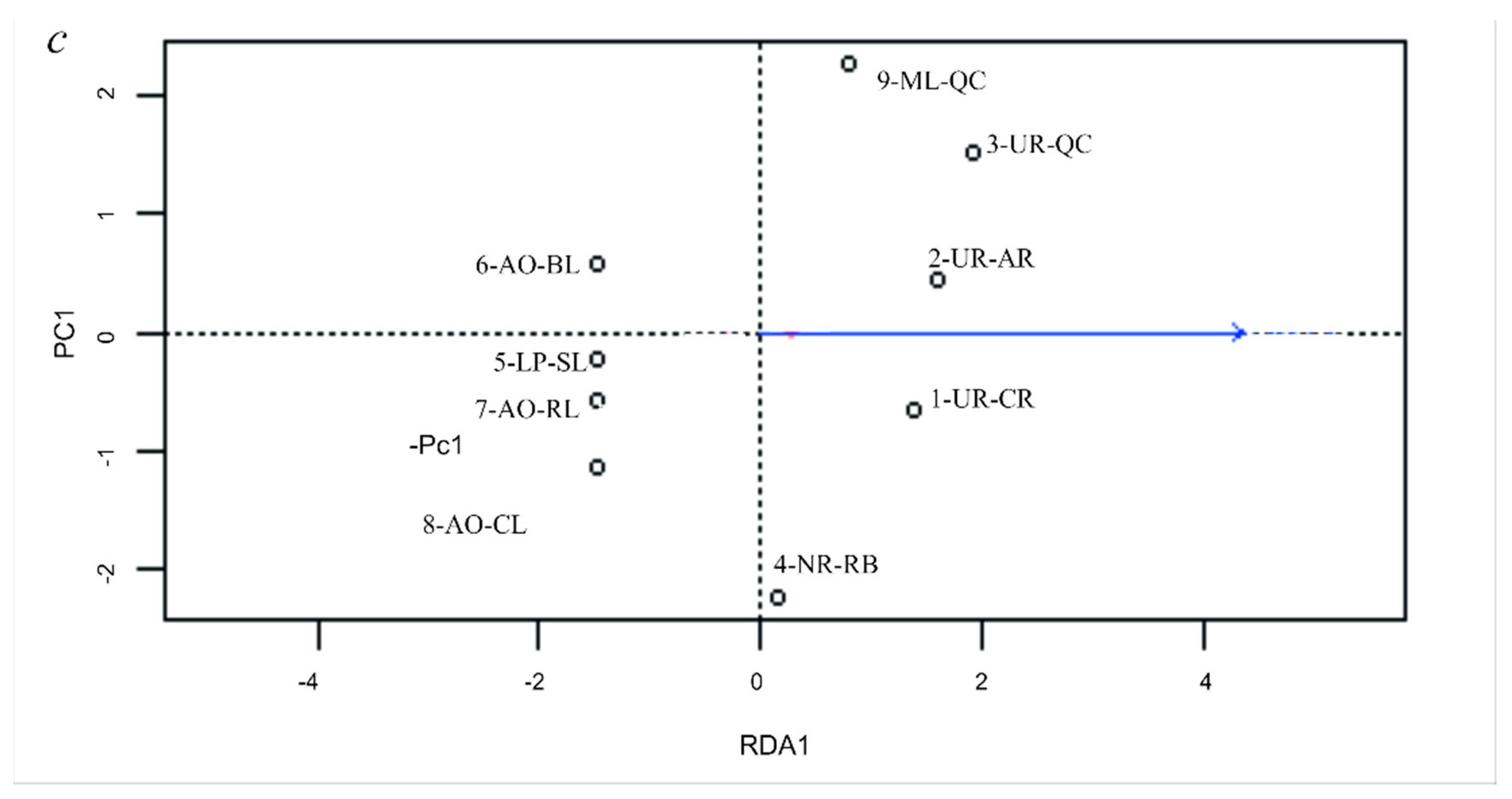
3.5. Genetic Differentiation by Environmental Factors
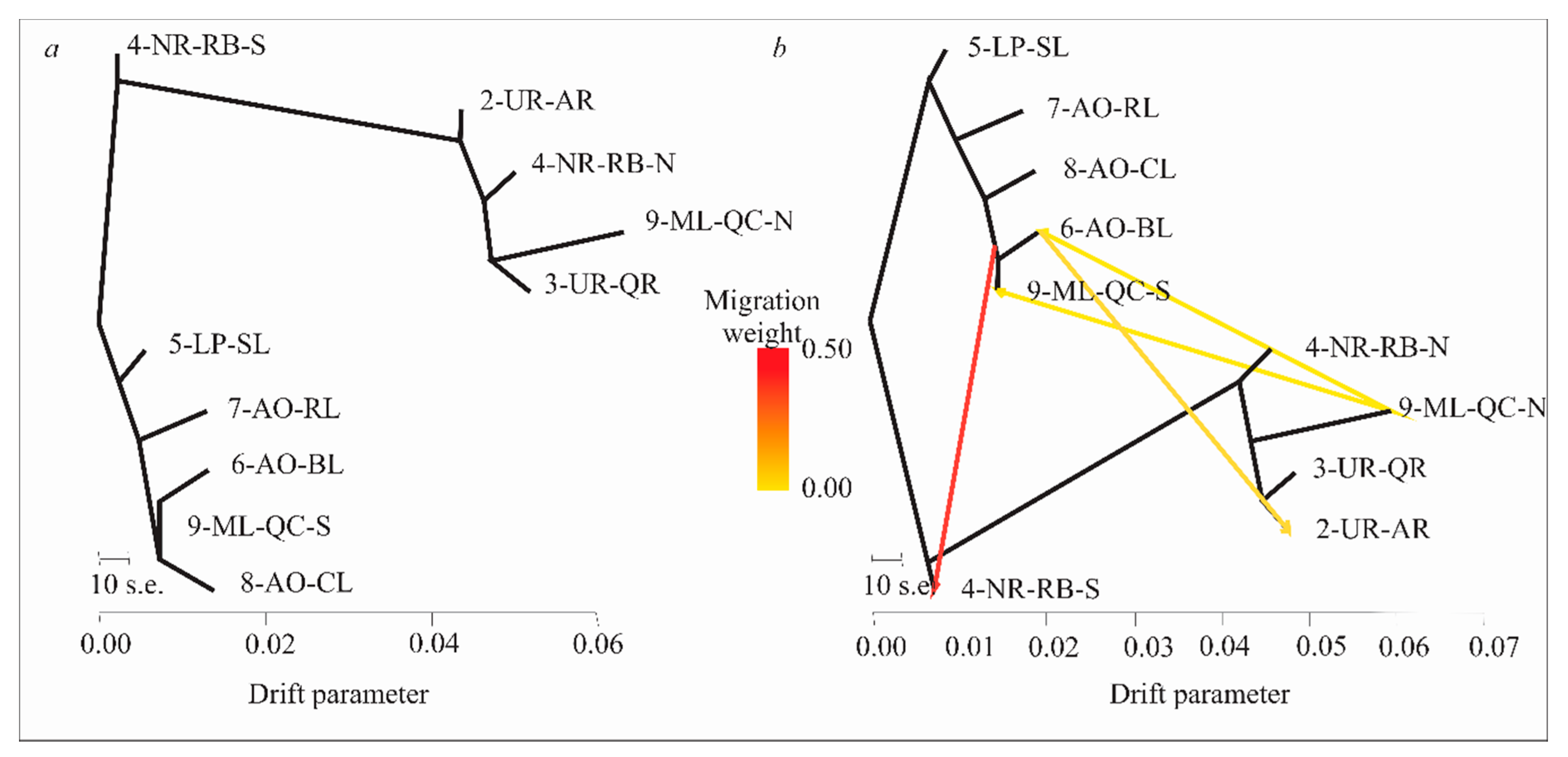
3.6. Evolutionary History of Population
4. Discussion
4.1. Wide Distribution of Interbred mtDNA Lineages
4.2. Genomic Pattern of Differentiation
4.3. Genetic Substructure within Genomic Clusters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hubert, N.; Renno, J.F. Historical biogeography of South American freshwater fishes. J. Biogeogr. 2006, 33, 1414–1436. [Google Scholar]
- Lundberg, J.G.; Marshall, L.G.; Guerrero, J.; Horton, B.; Malabarba, M.C.; Wesselingh, F. The stage for neotropical fish diversification: A history of tropical South American rivers. Phylogeny Classif. Neotrop. Fishes 1998, 603, 14–48. [Google Scholar]
- Albert, J.S.; Reis, R.E. Introduction to Neotropical Freshwaters. In Historical Biogeography of Neotropical Freshwater Fishes; Albert, J.S., Reis, R.E., Eds.; University of California Press: Berkeley, CA, USA, 2011; pp. 3–19. [Google Scholar]
- Wilkinson, J.M.; Marshall, L.G.; Lundberg, J.G. River behavior on megafans and potential influences on diversification and distribution of aquatic organisms. J. S. Am. Earth Sci. 2006, 21, 151–172. [Google Scholar] [CrossRef]
- Zemlak, T.S.; Habit, E.M.; Walde, S.J.; Carrea, C.; Ruzzante, D.E. Surviving historical Patagonian landscapes and climate: Molecular insights from Galaxias maculatus. BMC Evol. Biol. 2010, 10, 67. [Google Scholar]
- Zemlak, T.S.; Walde, S.J.; Habit, E.M.; Ruzzante, D.E. Climate-Induced changes to the ancestral population size of two Patagonian galaxiids: The influence of glacial cycling. Mol. Ecol. 2011, 20, 5280–5294. [Google Scholar] [CrossRef]
- Ruzzante, D.E.; Walde, S.J.; Macchi, P.J.; Alonso, M.; Barriga, J.P. Phylogeography and phenotypic diversification in the Patagonian fish Percichthys trucha: The roles of Quaternary glacial cycles and natural selection. Biol. J. Linn. Soc. 2011, 103, 514–529. [Google Scholar]
- Perdices, A.; Bermingham, E.; Montilla, A.; Doadrio, I. Evolutionary history of the genus Rhamdia (Teleostei: Pimelodidae) in Central America. Mol. Phylogenet. Evol. 2002, 25, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Ribolli, J.; Zaniboni-Filho, E. Individual contributions to pooled-milt fertilizations of silver catfish Rhamdia quelen. Neotrop. Ichthyol. 2009, 7, 629–634. [Google Scholar]
- Ribolli, J.; Scaranto, B.M.; Shibatta, O.A.; Bombardelli, R.A.; Zaniboni-Filho, E. DNA barcoding confirms the occurrence of Rhamdia branneri and Rhamdia voulezi (Siluriformes: Heptapteridae) in the Iguaçu River Basin. Neotrop. Ichthyol. 2017, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Scaranto, B.M.S.; Ribolli, J.; Zaniboni-Filho, E. DNA barcoding reveals blend of silver catfish Rhamdia species from fish farms in Southern Brazil. Aquac. Res. 2018, 49, 1907–1913. [Google Scholar] [CrossRef]
- Loureiro, M.; Zarucki, M.; González, I.; Vidab, N.; Fabiano, G. Peces Continentales. In Especies Prioritarias Para La Conservación En Uruguay. Vertebrados, Moluscos Continentales Y Plantas Vasculares; Soutullo, A., Clavijo, C.M.-L.J.A., Eds.; SNAP/DINAMA/MVOTMA y DICYT/MEC: Montevideo, Uruguay, 2013; pp. 91–112. ISBN 9789974825970. [Google Scholar]
- Silfvergrip, A. A Systematic Revision of the Neotropical Catfish Genus Rhamdia (Teleostei, Pimelodidae). Ph.D. Thesis, Swedish Museum of Natural History, Stockholm, Sweden, 1996. [Google Scholar]
- Ríos, N.; Bouza, C.; Gutiérrez, V.; García, G. Species complex delimitation and patterns of population structure at different geographic scales in Neotropical silver catfish (Rhamdia: Heptapteridae). Environ. Biol. Fishes 2017, 100, 1–21. [Google Scholar] [CrossRef]
- Usso, M.C.; Santos, A.R.D.; Gouveia, J.G.; Frantine-Silva, W.; Araya-Jaime, C.; de Oliveira, M.L.M.; Foresti, F.; Giuliano-Caetano, L.; Dias, A.L. Genetic and Chromosomal Differentiation of Rhamdia quelen (Siluriformes, Heptapteridae) Revealed by Repetitive Molecular Markers and DNA Barcoding. Zebrafish 2019, 16, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angrizani, R.C.; Malabarba, L.R. Morphology and molecular data reveal the presence of two new species under Rhamdia quelen (Quoy & Gaimard, 1824) (Siluriformes: Heptapteridae) species complex. Zootaxa 2018, 4388, 41–60. [Google Scholar]
- Ríos, N.; Bouza, C.; García, G. Past hybridisation and introgression erased traces of mitochondrial lineages evolution in the Neotropical silver catfish Rhamdia quelen (Siluriformes: Heptapteridae). Hydrobiologia 2019, 830, 161–177. [Google Scholar] [CrossRef]
- Guo, B.; DeFaveri, J.; Sotelo, G.; Nair, A.; Merilä, J. Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks. BMC Biol. 2015, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Robledo, D.; Palaiokostas, C.; Bargelloni, L.; Martínez, P.; Houston, R. Applications of genotyping by sequencing in aquaculture breeding and genetics. Rev. Aquac. 2017, 1–13. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, W.; Guo, H.; Chen, B.; Zhou, Z.; Xu, J.; Zhang, D.; Xu, P. Population Genomics Reveals Genetic Divergence and Adaptive Differentiation of Chinese Sea Bass (Lateolabrax maculatus). Mar. Biotechnol. 2018, 20, 45–59. [Google Scholar] [CrossRef]
- Funk, W.C.; Mckay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Do Prado, F.D.; Vera, M.; Hermida, M.; Bouza, C.; Pardo, B.G.; Vilas, R.; Blanco, A.; Fernández, C.; Maroso, F.; Maes, G.E.; et al. Parallel evolution and adaptation to environmental factors in a marine flatfish: Implications for fisheries and aquaculture management of the turbot (Scophthalmus maximus). Evol. Appl. 2018, 11, 1322–1341. [Google Scholar] [CrossRef] [Green Version]
- Batista, P.D.; Janes, J.K.; Boone, C.K.; Murray, B.W.; Sperling, F.A.H. Adaptive and neutral markers both show continent-wide population structure of mountain pine beetle (Dendroctonus ponderosae). Ecol. Evol. 2016, 6, 6292–6300. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, S. Genomic rearrangements and the evolution of clusters of locally adaptive loci. Proc. Nat. Acad. Sci. USA 2013, 110, E1743–E1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feder, J.L.; Nosil, P. The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution (N. Y.) 2010, 64, 1729–1747. [Google Scholar] [CrossRef]
- Feder, J.L.; Egan, S.P.; Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Via, S. Divergence hitchhiking and the spread of genomic isolation during ecological speciation-with-gene-flow. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Gagnaire, P.A.; Pavey, S.A.; Normandeau, E.; Bernatchez, L. The genetic architecture of reproductive isolation during speciation-with-gene-flow in lake whitefish species pairs assessed by rad sequencing. Evolution (N. Y.) 2013, 67, 2483–2497. [Google Scholar] [CrossRef]
- Bernal, M.A.; Gaither, M.R.; Simison, W.B.; Rocha, L.A. Introgression and selection shaped the evolutionary history of sympatric sister-species of coral reef fishes (genus: Haemulon). Mol. Ecol. 2017, 26, 639–652. [Google Scholar] [CrossRef]
- Zhong, Y.; Yang, A.; Liu, S.; Liu, L.; Li, Y.; Wu, Z.; Yu, F. RAD-Seq data point to a distinct split in Liriodendron (Magnoliaceae) and obvious east-west genetic divergence in L. chinense. Forests 2018, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Fenerich, P.C.; Foresti, F.; Oliveira, C. Nuclear DNA content in 20 species of Siluriformes (Teleostei: Ostariophysi) from the Neotropical region. Genet. Mol. Biol. 2004, 27, 350–354. [Google Scholar] [CrossRef]
- Medrano, J.F.; Aasen, E.; Sharrow, L. DNA extraction from nucleated red blood cells. Biotechniques 1990, 8, 43. [Google Scholar]
- Palumbi, S.; Martin, A.; Romano, S.; McMillam, W.O.; Stice, L.; Grabowski, G. The Simple Fools Guide to PCR; Department of Zoology and Kewalo Marine Laboratory: Honolulu, HI, USA, 1991. [Google Scholar]
- Vergara, J.; de las Mercedes Azpelicueta, M.; Garcia, G. Phylogeography of the Neotropical catfish Pimelodus albicans (Siluriformes: Pimelodidae) from río de la Plata basin, South America, and conservation. Neotrop. Ichthyol. 2008, 6, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.L.; Ortega-Lara, A.; Sánchez-Garcés, G.C.; Alford, M.H. Genetic and Morphometric Evidence for the Recognition of Several Recently Synonymized Species of Trans-Andean Rhamdia (Pisces: Siluriformes: Heptapteridae). Copeia 2015, 103, 563–579. [Google Scholar] [CrossRef]
- Rosso, J.J.; Mabragaña, E.; González Castro, M.; Díaz de Astarloa, J.M. DNA barcoding Neotropical fishes: Recent advances from the Pampa Plain, Argentina. Mol. Ecol. Resour. 2012, 12, 999–1011. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Sugiura, N. Further Analysis of the Data by Anaike’ S Information Criterion and the Finite Corrections. Commun. Stat. Theory Methods 1978, 7, 13–26. [Google Scholar] [CrossRef]
- Minin, V.; Abdo, Z.; Joyce, P.; Sullivan, J. Performance-Based Selection of Likelihood Models for Phylogeny Estimation. Syst. Biol. 2003, 52, 674–683. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mckay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef]
- Maroso, F.; Hermida, M.; Millán, A.; Blanco, A.; Saura, M.; Fernández, A.; Dalla Rovere, G.; Bargelloni, L.; Cabaleiro, S.; Villanueva, B.; et al. Highly dense linkage maps from 31 full-sibling families of turbot (Scophthalmus maximus) provide insights into recombination patterns and chromosome rearrangements throughout a newly refined genome assembly. DNA Res. 2018, 25, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics, FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 June 2018).
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tools | Calling genotypes from 2bRAD sequence data v2.0. Available online: http://people.oregonstate.edu/~meyere/2bRAD_analysis2.1.html (accessed on 28 June 2018).
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genet. Soc. Am. 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Narum, S.R.; Hess, J.E. Comparison of FST outlier tests for SNP loci under selection. Mol. Ecol. Resour. 2011, 11, 184–194. [Google Scholar] [CrossRef]
- De Mita, S.; Thuillet, A.C.; Gay, L.; Ahmadi, N.; Manel, S.; Ronfort, J.; Vigouroux, Y. Detecting selection along environmental gradients: Analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol. Ecol. 2013, 22, 1383–1399. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, S.; Yao, J.; Bao, L.; Zhang, J.; Li, Y.; Jiang, C.; Sun, L.; Wang, R.; Zhang, Y.; et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database (Oxf.) 2018. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal annotation and visualization tool in functional genomics research. Application note. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Malinsky, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and fineRADstructure: Population Inference from RADseq Data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar] [CrossRef]
- Klimova, A.; Ortega-Rubio, A.; Vendrami, D.L.J.; Hoffman, J.I. Genotyping by sequencing reveals contrasting patterns of population structure, ecologically mediated divergence, and long-distance dispersal in North American palms. Ecol. Evol. 2018, 8, 5873–5890. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- MVOTMA: Sistema de Información Ambiental. Available online: https://www.dinama.gub.uy/visualizador/index.php?vis=sig (accessed on 10 June 2019).
- Iglesias, C.; Mazzeo, N.; Goyenola, G.; Fosalba, C.; Teixeira De Mello, F.; García, S.; Jeppesen, E. Field and experimental evidence of the effect of Jenynsia multidentata, a small omnivorous-planktivorous fish, on the size distribution of zooplankton in subtropical lakes. Freshw. Biol. 2008, 53, 1797–1807. [Google Scholar] [CrossRef]
- Vidal, L.; Rodríguez-Gallego, L.; Conde, D.; Martínez-López, W.; Bonilla, S. Biomass of autotrophic picoplankton in subtropical coastal lagoons: Is it relevant? Limnetica 2007, 26, 441–452. [Google Scholar]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan Community Ecology Package: Ordination Methods, Diversity Analysis and Other Functions for Community and Vegetation Ecologists. 2015. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 10 January 2019).
- Pickrell, J.K.; Pritchard, J.K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. aki Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Carvalho, T.P.; Albert, J.S. The amazon-paraguay divide. In Historical Biogeography of Neotropical Freshwater Fishes; Albert, J.S., Reis, R.E., Eds.; University of California Press: Berkeley, CA, USA, 2011; pp. 193–202. [Google Scholar]
- Hubert, N.; Duponchelle, F.; Nuñez, J.; Garcia-Davila, C.; Paugy, D.; Renno, J.F. Phylogeography of the piranha genera Serrasalmus and Pygocentrus: Implications for the diversification of the Neotropical ichthyofauna. Mol. Ecol. 2007, 16, 2115–2136. [Google Scholar] [CrossRef]
- Ribeiro, A.; Jacob, R.; Silva, R.; Lima, F.C.T.; Ferreira, D.C.; Ferreira, K.M.; Mariguela, T.C.; Pereira, L.H.G.; Oliveira, C. Distributions and phylogeographic data of rheophilic freshwater fishes provide evidences on the geographic extension of a central-brazilian amazonian. Neotrop. Ichthyol. 2013, 11, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Garavello, J.C.; Shibatta, O.A. Reappraisal of Rhamdia branneri Haseman, 1911 and R. voulezi Haseman, 1911 (Siluriformes: Heptapteridae) from the rio Iguaçu with notes on their morphometry and karyotype. Neotrop. Ichthyol. 2016, 14. [Google Scholar] [CrossRef] [Green Version]
- Renaut, S.; Rowe, H.C.; Ungerer, M.C.; Rieseberg, L.H. Genomics of homoploid hybrid speciation: Diversity and transcriptional activity of long terminal repeat retrotransposons in hybrid sunflowers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffery, N.W.; Stanley, R.R.; Wringe, B.F.; Guijarro-Sabaniel, J.; Bourret, V.; Bernatchez, L.; Bradbury, I.R. Range-wide parallel climate-associated genomic clines in Atlantic salmon. R. Soc. Open Sci. 2017, 4, 171394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, S.L.; Hamilton, D.; Currie, S. A comparison of the heat shock response in juvenile and adult rainbow trout (Oncorhynchus mykiss)—Implications for increased thermal sensitivity with age. Can. J. Fish. Aquat. Sci. 2009, 66, 91–100. [Google Scholar] [CrossRef]
- Bradbury, I.R.; Hubert, S.; Higgins, B.; Borza, T.; Bowman, S.; Paterson, I.G.; Snelgrove, P.V.; Morris, C.J.; Gregory, R.S.; Hardie, D.C.; et al. Parallel adaptive evolution of Atlantic cod on both sides of the Atlantic Ocean in response to temperature. Proc. R. Soc. B Biol. Sci. R. Soc. 2010, 277, 3725–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panario, D.; Gutiérrez, O. Introducción a la geomorfología de lagunas costeras, lagos someros y charcas de Uruguay. In El Holoceno en la Zona costera de Uruguay; Ediciones Universitarias: Montevideo, Uruguay, 2011; pp. 49–63. [Google Scholar]
- Ubilla, M. Mammalian biostratigraphy of Pleistocene fluvial deposits in northern Uruguay, South America. Proc. Geol. Assoc. 2004, 115, 347–357. [Google Scholar] [CrossRef]
- Abram, Q.H.; Dixon, B.; Katzenback, B.A. Impacts of Low Temperature on the Teleost Immune System. Biology 2017, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakon, H.H. Adaptive evolution of voltage-gated sodium channels: The first 800 million years. Proc. Nat. Acad. Sci. USA 2012, 109, 10619–10625. [Google Scholar] [CrossRef] [Green Version]
- Berg, P.R.; Jentoft, S.; Star, B.; Ring, K.H.; Knutsen, H.; Lien, S.; Jakobsen, K.S.; André, C. Adaptation to low salinity promotes genomic divergence in Atlantic Cod (Gadus morhua L.). Genome Biol. Evol. 2015, 7, 1644–1663. [Google Scholar] [CrossRef]
- Defaveri, J.; Merilä, J. Local adaptation to salinity in the three-spined stickleback? J. Evol. Biol. 2014, 27, 290–302. [Google Scholar] [CrossRef]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.P.; Bank, C.; Brännström, Å.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, N.J.; Martin, S.H.; Kozak, K.M.; Salazar, C.; Dasmahapatra, K.K.; Davey, J.W.; Baxter, S.W.; Blaxter, M.L.; Mallet, J.; Jiggins, C.D. Genome-wide patterns of divergence and gene flow across a butterfly radiation. Mol. Ecol. 2013, 22, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.L.; Rieseberg, L.H. Divergence is focused on few genomic regions early in speciation: Incipient speciation of sunflower ecotypes. Evolution (N. Y.) 2013, 67, 2468–2482. [Google Scholar] [CrossRef] [PubMed]
- Stölting, K.N.; Nipper, R.; Lindtke, D.; Caseys, C.; Waeber, S.; Castiglione, S.; Lexer, C. Genomic scan for single nucleotide polymorphisms reveals patterns of divergence and gene flow between ecologically divergent species. Mol. Ecol. 2013, 22, 842–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deagle, B.E.; Jones, F.C.; Chan, Y.F.; Absher, D.M.; Kingsley, D.M.; Reimchen, T.E. Population genomics of parallel phenotypic evolution in stickleback across stream-lake ecological transitions. Proc. R. Soc. B Biol. Sci. 2012, 279, 1277–1286. [Google Scholar] [CrossRef]
- Kirkpatrick, M.; Kern, A. Where’s the Money? Inversions, Genes, and the Hunt for Genomic Targets of Selection. Genetics 2012, 190, 1153–1155. [Google Scholar] [CrossRef] [Green Version]
- Faria, R.; Chaube, P.; Morales, H.E.; Larsson, T.; Lemmon, A.R.; Lemmon, E.M.; Rafajlović, M.; Panova, M.; Ravinet, M.; Johannesson, K.; et al. Multiple chromosomal rearrangements in a hybrid zone between Littorina saxatilis ecotypes. Mol. Ecol. 2019, 28, 1375–1393. [Google Scholar] [CrossRef] [Green Version]
- Jones, F.C.; Chan, Y.F.; Schmutz, J.; Grimwood, J.; Brady, S.D.; Southwick, A.M.; Absher, D.M.; Myers, R.M.; Reimchen, T.E.; Deagle, B.E.; et al. A genome-wide SNP genotyping array reveals patterns of global and repeated species-pair divergence in sticklebacks. Curr. Biol. 2012, 22, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Berg, P.R.; Star, B.; Pampoulie, C.; Bradbury, I.R.; Bentzen, P.; Hutchings, J.A.; Jentoft, S.; Jakobsen, K.S. Trans-oceanic genomic divergence of Atlantic cod ecotypes is associated with large inversions. Heredity 2017, 119, 418. [Google Scholar] [CrossRef] [Green Version]
- Lorenzetti, D.; Poirier, C.; Zhao, M.; Overbeek, P.A.; Harrison, W.; Bishop, C.E. A transgenic insertion on mouse chromosome 17 inactivates a novel immunoglobulin superfamily gene potentially involved in sperm-egg fusion. Mamm. Genome 2014, 25, 141–148. [Google Scholar] [CrossRef]
- Quagio-Grassiotto, I.; Spadella, M.A.; de Carvalho, M.; Oliveira, C. Comparative description and discussion of spermiogenesis and spermatozoal ultrastructure in some species of Heptapteridae and Pseudopimelodidae. Neotrop. Ichthyol. 2005, 3, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Quagio-Grassiotto, I.; Oliveira, C. Sperm ultrastructure and a new type of spermiogenesis in two species of Pimelodidae, with a comparative review of sperm ultrastructure in Siluriformes (Teleostei: Ostariophysi). Zool. Anz. 2008, 247, 55–66. [Google Scholar] [CrossRef]
- Bird, C.E.; Fernandez-Silva, I.; Skillings, D.J.; Toonen, R.J. Shared genomic outliers across two divergent population clusters of a highly threatened seagrass View project Vulnerability of Hawaiian tree snails to climate change, and the potential for adaptation View project. Evol. Biol. 2012, 39, 158–180. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | N RAD-seq | N cytb | N COI |
|---|---|---|---|
| 1-UR-CR | 2 | 7 (1) | 2 |
| 2-UR-AR | 7 | 11 (0) | 3 |
| 3-UR-QR | 4 | 6 (0) | - |
| 4-NR-RB | 10 | 23 (0) | 1 |
| 5-LP-SL | 10 | 49 (3) | - |
| 6-AO-BL | 9 | 19 (9) | - |
| 7-AO-RL | 9 | 17 (1) | 1 |
| 8-AO-CL | 10 | 36 (6) | 1 |
| 9-ML-QC | 10 | 16 (2) | 1 |
| 10-VC | 3 | 12 (0) | 3 |
| SNP | Homologous Sequence (E-Value) | Gene | Gene Mining | Biological Processes of Gene Mining |
|---|---|---|---|---|
| 64979_10 | Ictalurus punctatus genome Chr 5: 13,660,451–13,660,486 (9 e−10) | Unannotated sequence | zbtb34 angptl2 ralgps1 | RNA polymerase II regulatory region; DNA-binding transcription repressor immune; angiogenesis; hematopoiesis; cell differentiation; positive regulation of Notch signaling pathway; signal transduction (GTPase, Ral protein); cell differentiation; anatomical structure and morphogenesis; embryo development; cellular nitrogen compound metabolic process |
| 15866_28 | I. punctatus genome Chr 5: 13,807,353–13,807,388 (5 e−5) | ralgps1 | ||
| 49857_34 | I. punctatus genome Chr 8: 7,712,981–7,713,013 (2 e−10) | kdrl | kctd8 lnx2 loxl3 trmt112 atg2a itpkb tc1a seipin-like cdx1; slc25a6 ant3ANT3 | Protein binding; scavenger receptor activity; copper ion binding; oxidoreductase activity; protein heterodimerization activity; kinase activity; DNA binding; protein kinase and protein tyrosine kinase activity; ATP binding and transmembrane transporter activity; DNA binding; regulation of transcription, DNA-templated; multicellular organism development |
| 9230_26 | XM_027135514.1 I. punctatus genome Chr 8: 7,757,023–7,757,984 (7 e−5) | chic1 | ||
| 449_34 | XM_027165565.1 I. punctatus genome Chr 8: 8,029,587–8,031,148 (4 e−7) | fbxw2 |
| Locality | N | Na | HE | FIS |
|---|---|---|---|---|
| 2-UR-AR | 7 | 1.47 | 0.276 (0.163) | −0.018 |
| 4-NR-RB | 10 | 1.60 | 0.283 (0.158) | 0.285 * |
| 5-LP-SL | 10 | 1.36 | 0.249 (0.148) | 0.013 |
| 6-AO-BL | 9 | 1.20 | 0.328 (0.139) | −0.015 |
| 7-AO-RL | 9 | 1.22 | 0.268 (0.154) | 0.010 |
| 8-AO-CL | 10 | 1.24 | 0.277 (0.151) | 0.000 |
| 9-ML-QC | 10 | 1.37 | 0.324 (0.154) | 0.486 * |
| 1-UR-CR | 2-UR-AR | 3-UR-UQ | 4-NR-RB | 9-ML-QC | 5-LP-SL | 6-AO-BL | 7-AO-RL | 8-AO-CL | 10-VC | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Riverine | North | 1-UR-CR | 0.000 | |||||||||
| 2-UR-AR | 0.036 | 0.000 | ||||||||||
| 3-UR-UQ | 0.075 | 0.040 | 0.000 | |||||||||
| Centre | 4-NR-RB | 0.077 | 0.107 | 0.143 | 0.000 | |||||||
| 9-ML-QC | 0.121 | 0.098 | 0.151 | 0.098 | 0.000 | |||||||
| Coastal lagoon | South | 5-LP-SL | 0.458 | 0.447 | 0.519 | 0.210 | 0.383 | 0.000 | ||||
| 6-AO-BL | 0.537 | 0.496 | 0.582 | 0.261 | 0.427 | 0.196 | 0.000 | |||||
| 7-AO-RL | 0.537 | 0.497 | 0.581 | 0.265 | 0.432 | 0.174 | 0.246 | 0.000 | ||||
| 8-AO-CL | 0.555 | 0.516 | 0.597 | 0.280 | 0.445 | 0.209 | 0.213 | 0.250 | 0.000 | |||
| Hatchery | 10-VC | 0.189 | 0.292 | 0.342 | 0.116 | 0.269 | 0.163 | 0.276 | 0.270 | 0.307 | 0.000 | |
| 1-UR-CR | 2-UR-AR | 3-UR-UQ | 4-NR-RB | 9-ML-QC | 5-LP-SL | 6-AO-BL | 7-AO-RL | 8-AO-CL | 10-VC | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Riverine | North | 1-UR-CR | 0.000 | |||||||||
| 2-UR-AR | 0.020 | 0.000 | ||||||||||
| 3-UR-UQ | 0.045 | 0.032 | 0.000 | |||||||||
| Centre | 4-NR-RB | 0.051 | 0.058 | 0.076 | 0.000 | |||||||
| 9-ML-QC | 0.126 | 0.081 | 0.129 | 0.081 | 0.000 | |||||||
| Coastal lagoon | South | 5-LP-SL | 0.270 | 0.258 | 0.307 | 0.138 | 0.252 | 0.000 | ||||
| 6-AO-BL | 0.352 | 0.309 | 0.380 | 0.186 | 0.300 | 0.170 | 0.000 | |||||
| 7-AO-RL | 0.358 | 0.310 | 0.379 | 0.191 | 0.308 | 0.144 | 0.207 | 0.000 | ||||
| 8-AO-CL | 0.378 | 0.334 | 0.399 | 0.203 | 0.316 | 0.173 | 0.185 | 0.202 | 0.000 | |||
| Hatchery | 10-VC | 0.065 | 0.173 | 0.198 | 0.110 | 0.221 | 0.142 | 0.239 | 0.240 | 0.269 | 0.000 | |
| 1-UR-CR | 2-UR-AR | 3-UR-UQ | 4-NR-RB | 9-ML-QC | 5-LP-SL | 6-AO-BL | 7-AO-RL | 8-AO-CL | 10-VC | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Riverine | North | 1-UR-CR | 0.000 | |||||||||
| 2-UR-AR | 0.120 | 0.000 | ||||||||||
| 3-UR-UQ | 0.372 | −0.001 | 0.000 | |||||||||
| Centre | 4-NR-RB | 0.152 | 0.307 | 0.390 | 0.000 | |||||||
| 9-ML-QC | 0.035 | 0.149 | 0.239 | 0.048 | 0.000 | |||||||
| Coastal lagoon | South | 5-LP-SL | 0.947 | 0.916 | 0.999 | 0.454 | 0.668 | 0.000 | ||||
| 6-AO-BL | 0.942 | 0.910 | 0.999 | 0.437 | 0.655 | 0.000 | 0.000 | |||||
| 7-AO-RL | 0.942 | 0.911 | 0.999 | 0.439 | 0.655 | 0.000 | 0.000 | 0.000 | ||||
| 8-AO-CL | 0.948 | 0.917 | 0.999 | 0.462 | 0.673 | 0.000 | 0.000 | 0.000 | 0.000 | |||
| Hatchery | 10-VC | 0.647 | 0.750 | 0.883 | 0.156 | 0.404 | 0.389 | 0.353 | 0.362 | 0.401 | 0.000 | |
| Total | Neutral | Outliers | ||
|---|---|---|---|---|
| RDA | p-Value | 0.007 | 0.003 | 0.006 |
| r2 | 0.576 | 0.517 | 0.827 | |
| Adjusted r2 | 0.515 | 0.448 | 0.802 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos, N.; Casanova, A.; Hermida, M.; Pardo, B.G.; Martínez, P.; Bouza, C.; García, G. Population Genomics in Rhamdia quelen (Heptapteridae, Siluriformes) Reveals Deep Divergence and Adaptation in the Neotropical Region. Genes 2020, 11, 109. https://doi.org/10.3390/genes11010109
Ríos N, Casanova A, Hermida M, Pardo BG, Martínez P, Bouza C, García G. Population Genomics in Rhamdia quelen (Heptapteridae, Siluriformes) Reveals Deep Divergence and Adaptation in the Neotropical Region. Genes. 2020; 11(1):109. https://doi.org/10.3390/genes11010109
Chicago/Turabian StyleRíos, Néstor, Adrián Casanova, Miguel Hermida, Belén G. Pardo, Paulino Martínez, Carmen Bouza, and Graciela García. 2020. "Population Genomics in Rhamdia quelen (Heptapteridae, Siluriformes) Reveals Deep Divergence and Adaptation in the Neotropical Region" Genes 11, no. 1: 109. https://doi.org/10.3390/genes11010109
APA StyleRíos, N., Casanova, A., Hermida, M., Pardo, B. G., Martínez, P., Bouza, C., & García, G. (2020). Population Genomics in Rhamdia quelen (Heptapteridae, Siluriformes) Reveals Deep Divergence and Adaptation in the Neotropical Region. Genes, 11(1), 109. https://doi.org/10.3390/genes11010109