Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease


Abstract
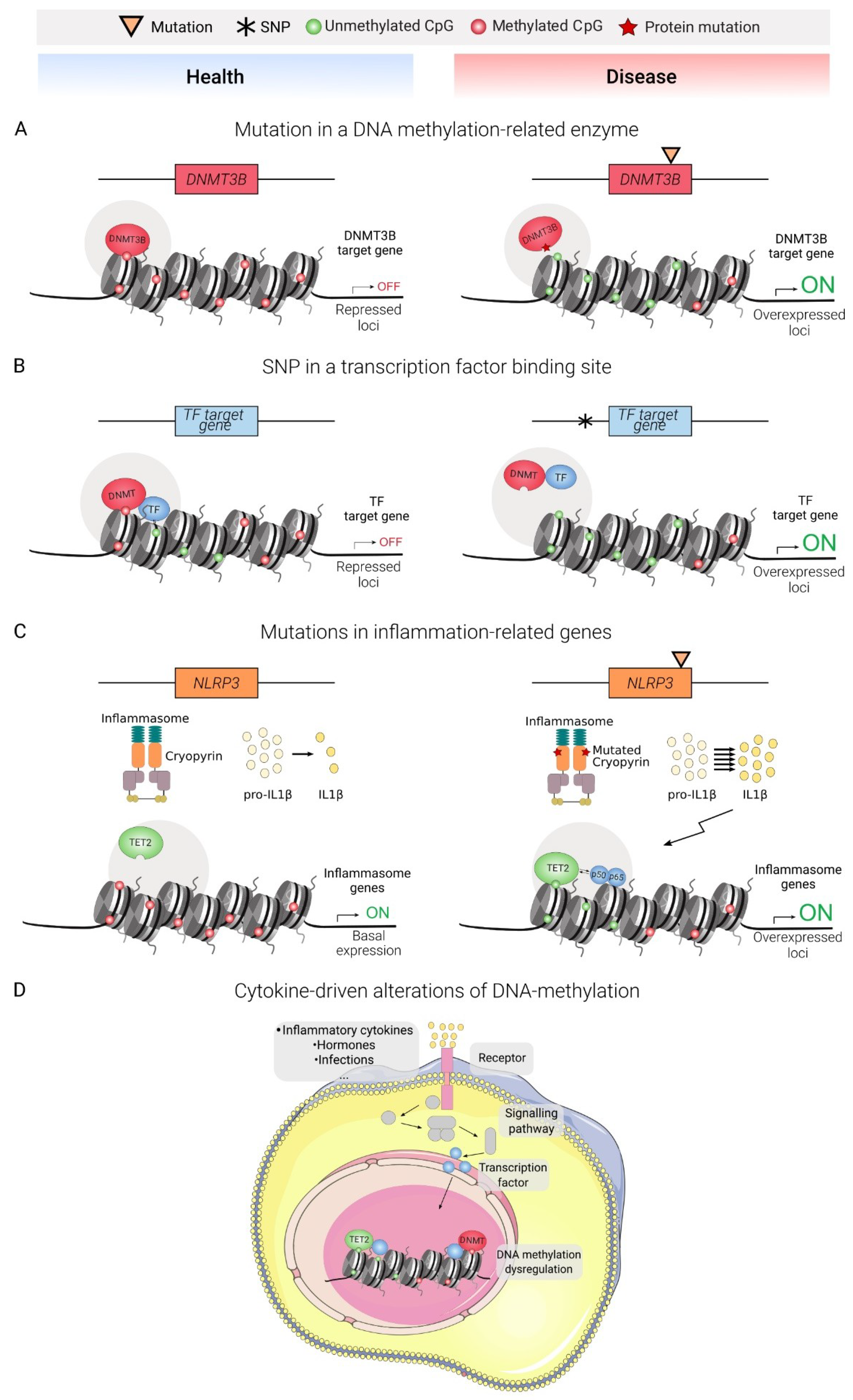
:1. Introduction: DNA Methylation Is a Cornerstone Regulator in Health and Disease
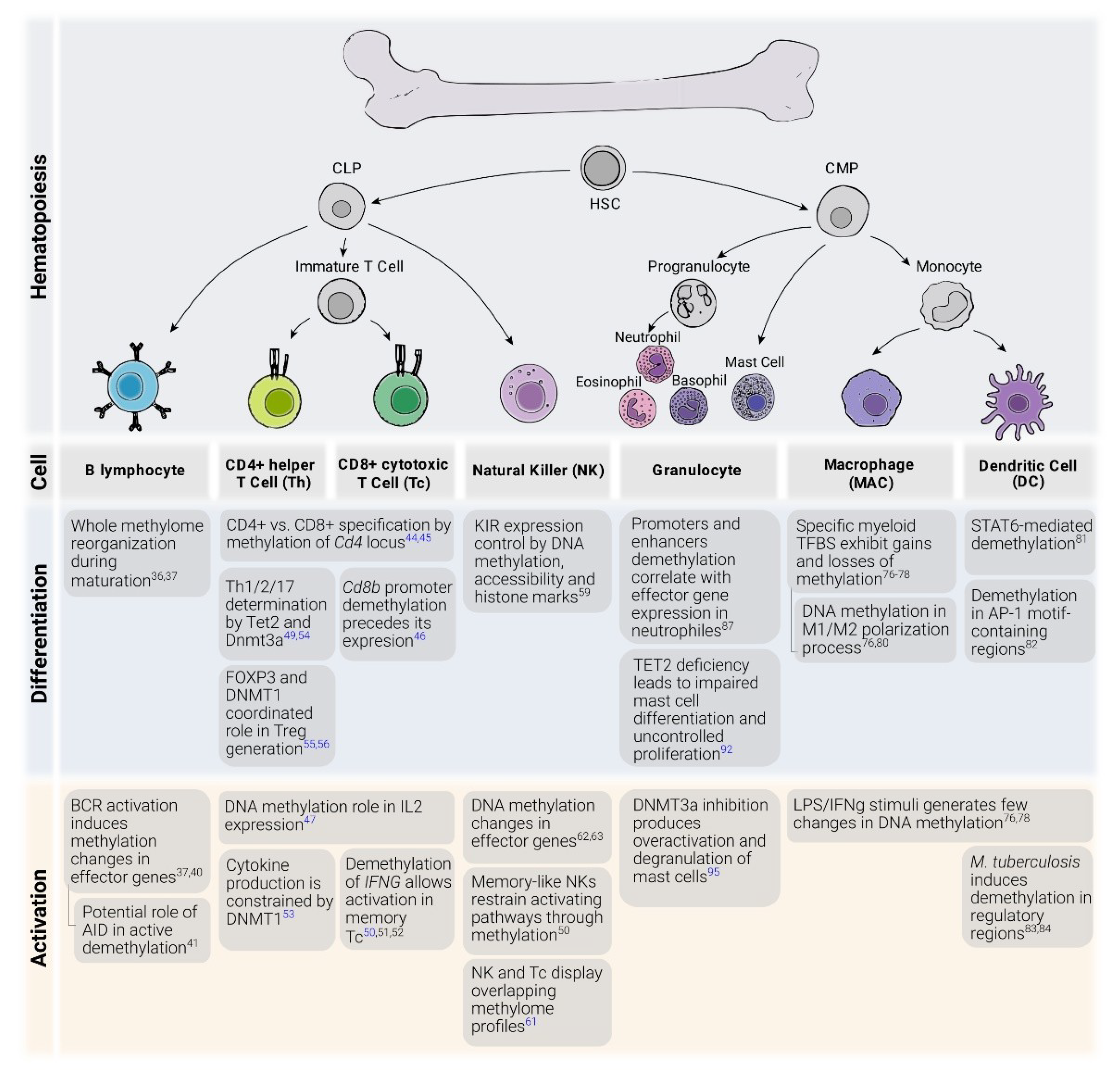
2. DNA Methylation in the Immune System
2.1. B Cells
2.2. T Cells
2.3. Innate Lymphoid Cells
2.4. Monocytes and Macrophages
2.5. Granulocytes
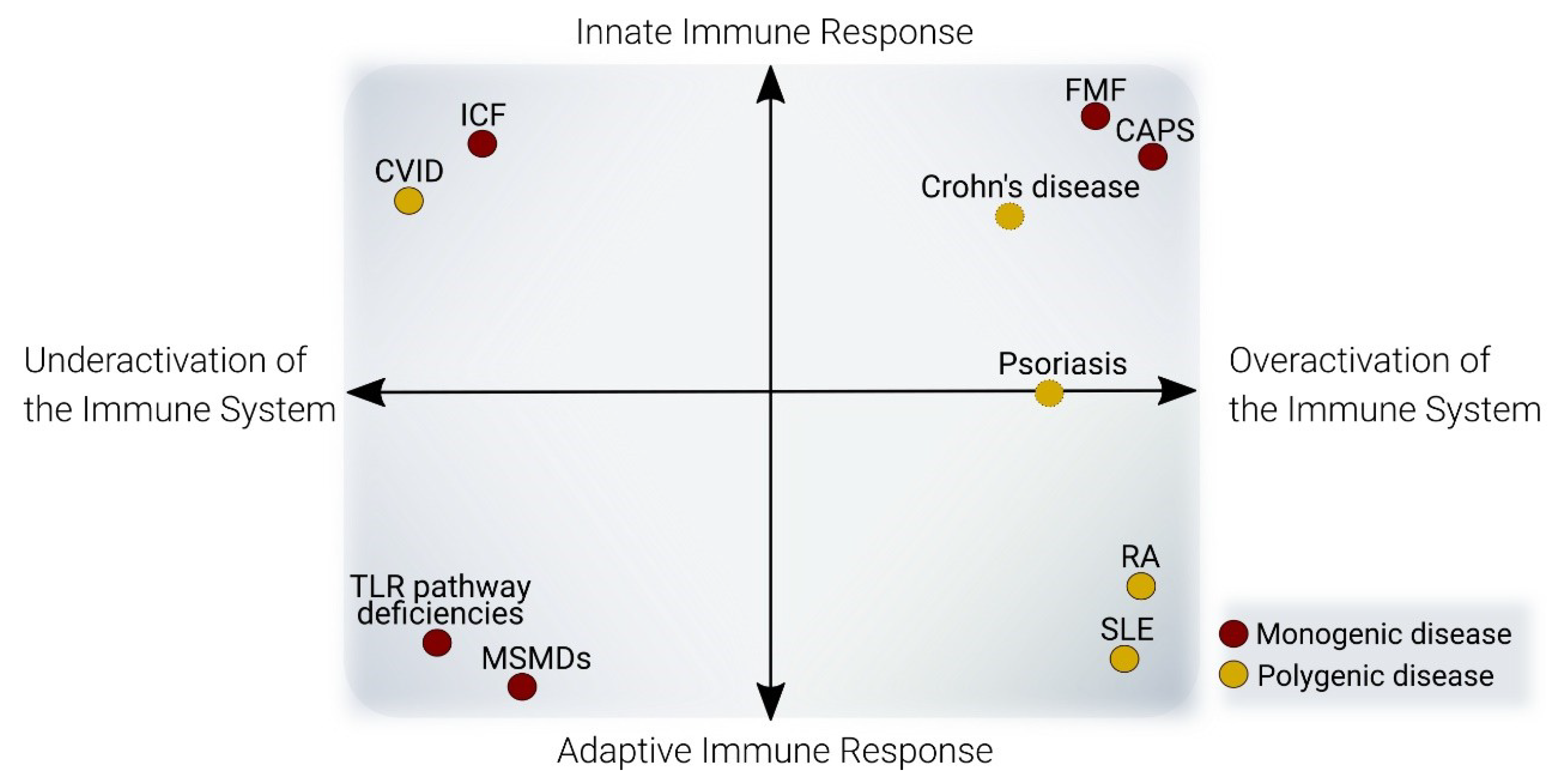
3. DNA Methylation in Immune-Mediated Diseases
3.1. DNA Methylation Defects in Conditions with Overactivation of the Immune System
3.1.1. Autoimmune Diseases
Rheumatoid Arthritis
Systemic Lupus Erythematosus
3.1.2. Autoinflammatory Diseases
Cryopyrin-Associated Periodic Fever Syndrome
Familial Mediterranean Fever
3.2. DNA Methylation Defects in Conditions with a Deficient Immune System
3.2.1. Adaptive Immune Deficiencies
Immunodeficiency, Centromeric Region Instability, and Facial Anomalies Syndrome
Common Variable Immunodeficiency
3.2.2. Innate Immune Deficiencies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Argelaguet, R.; Kapourani, C.A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. ScNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells e. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.R.; Urich, M.A.; Chen, H.; et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ge, B.; Casale, F.P.; Vasquez, L.; Kwan, T.; Garrido-Martín, D.; Watt, S.; Yan, Y.; Kundu, K.; Ecker, S.; et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 2016, 167, 1398–1414.e24. [Google Scholar] [CrossRef] [Green Version]
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 2013, 155, 621–635. [Google Scholar] [CrossRef] [Green Version]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2012, 13, 7–13. [Google Scholar] [CrossRef]
- Ford, E.; Grimmer, M.R.; Stolzenburg, S.; Bogdanovic, O.; de Mendoza, A.; Farnham, P.J.; Blancafort, P.; Lister, R. Frequent lack of repressive capacity of promoter DNA methylation identified through genome-wide epigenomic manipulation. bioRxiv 2017, 170506. [Google Scholar]
- Korthauer, K.; Irizarry, R.A. Genome-wide repressive capacity of promoter DNA methylation is revealed through epigenomic manipulation. bioRxiv 2018, 381145. [Google Scholar]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; BourC’His, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Álvarez-Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Ehrlich, L.I.R.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Berest, I.; Keβler, S.; Nishimura, K.; Simón-Carrasco, L.; Vassiliou, G.S.; Pedersen, M.T.; Christensen, J.; Zaugg, J.B.; Helin, K. TET2 binding to enhancers facilitates transcription factor recruitment in hematopoietic cells. Genome Res. 2019, 29, 564–575. [Google Scholar] [CrossRef] [Green Version]
- Martin-Subero, J.I.; Oakes, C.C. Charting the dynamic epigenome during B-cell development. Semin. Cancer Biol. 2018, 51, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.Y.; Mav, D.; Shah, R.; Grimm, S.A.; Phadke, D.; Hatzi, K.; Melnick, A.; Geigerman, C.; Sobol, S.E.; Jaye, D.L.; et al. Dna methylation profiling in human b cells reveals immune regulatory elements and epigenetic plasticity at alu elements during b-cell activation. Genome Res. 2013, 23, 2030–2041. [Google Scholar] [CrossRef] [Green Version]
- Schuyler, R.P.; Merkel, A.; Raineri, E.; Altucci, L.; Vellenga, E.; Martens, J.H.A.; Pourfarzad, F.; Kuijpers, T.W.; Burden, F.; Farrow, S.; et al. Distinct Trends of DNA Methylation Patterning in the Innate and Adaptive Immune Systems. Cell Rep. 2016, 17, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Merkel, A.; Heath, S.; Queirós, A.C.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.M.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Oakes, C.C.; Seifert, M.; Assenov, Y.; Gu, L.; Przekopowitz, M.; Ruppert, A.S.; Wang, Q.; Imbusch, C.D.; Serva, A.; Brocks, D.; et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet. 2016, 48, 253–264. [Google Scholar] [CrossRef]
- Dominguez, P.M.; Teater, M.; Chambwe, N.; Kormaksson, M.; Redmond, D.; Ishii, J.; Vuong, B.; Chaudhuri, J.; Melnick, A.; Vasanthakumar, A.; et al. DNA Methylation Dynamics of Germinal Center B Cells Are Mediated by AID. Cell Rep. 2015, 12, 2086–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramiro, A.R.; Barreto, V.M. Activation-induced cytidine deaminase and active DNA demethylation. Trends Biochem. Sci. 2015, 40, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Catala-Moll, F.; Alvarez-Prado, A.; Rodriguez-Ubreva, J.; Abolhassani, H.; Martinez-Gallo, M.; Dieli-Crimi, R.; Soler-Palacin, P.; Kracker, S.; Hammarstrom, L.; Durandy, A.; et al. Activation-induced deaminase is critical for the establishment of DNA methylation patterns prior to the germinal center reaction. bioRxiv Immunol. 2019, in press. [Google Scholar]
- Sellars, M.; Huh, J.R.; Day, K.; Issuree, P.D.; Galan, C.; Gobeil, S.; Absher, D.; Green, M.R.; Littman, D.R. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat. Immunol. 2015, 16, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gialitakis, M.; Sellars, M.; Littman, D.R. The epigenetic landscape of lineage choice: Lessons from the heritability of Cd4 and Cd8 expression. In Current Topics in Microbiology and Immunology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 356, pp. 165–188. ISBN 9783642241024. [Google Scholar]
- Hamerman, J.A.; Page, S.T.; Pullen, A.M. Distinct methylation states of the CD8 beta gene in peripheral T cells and intraepithelial lymphocytes. J. Immunol. 1997, 159, 1240–1246. [Google Scholar] [PubMed]
- Bruniquel, D.; Schwartz, R.H. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat. Immunol. 2003, 4, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The Methylcytosine Dioxygenase Tet2 Promotes DNA Demethylation and Activation of Cytokine Gene Expression in T Cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Kersh, E.N.; Fitzpatrick, D.R.; Murali-Krishna, K.; Shires, J.; Speck, S.H.; Boss, J.M.; Ahmed, R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J. Immunol. 2006, 176, 4083–4093. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.M.; Suarez-Alvarez, B.; Lavín, J.L.; Mosén-Ansorena, D.; Baragaño Raneros, A.; Márquez-Kisinousky, L.; Aransay, A.M.; Lopez-Larrea, C. Epigenetic Networks Regulate the Transcriptional Program in Memory and Terminally Differentiated CD8 + T Cells. J. Immunol. 2017, 198, 937–949. [Google Scholar] [CrossRef] [Green Version]
- Abdelsamed, H.A.; Moustaki, A.; Fan, Y.; Dogra, P.; Ghoneim, H.E.; Zebley, C.C.; Triplett, B.M.; Sekaly, R.P.; Youngblood, B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med. 2017, 214, 1593–1606. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.P.; Fitzpatrick, D.R.; Beard, C.; Jessup, H.K.; Lehar, S.; Makar, K.W.; Pérez-Melgosa, M.; Sweetser, M.T.; Schlissel, M.S.; Nguyen, S.; et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001, 15, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Gamper, C.J.; Agoston, A.T.; Nelson, W.G.; Powell, J.D. Identification of DNA Methyltransferase 3a as a T Cell Receptor-Induced Regulator of Th1 and Th2 Differentiation. J. Immunol. 2009, 183, 2267–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Arvey, A.; Chinen, T.; Van Der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, Y.; Beier, U.H.; Han, R.; Bhatti, T.R.; Akimova, T.; Hancock, W.W. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood 2013, 121, 3631–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Santourlidis, S.; Graffmann, N.; Christ, J.; Uhrberg, M. Lineage-Specific Transition of Histone Signatures in the Killer Cell Ig-Like Receptor Locus from Hematopoietic Progenitor to NK Cells. J. Immunol. 2008, 180, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.J.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015, 42, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Luetke-Eversloh, M.; Cicek, B.B.; Siracusa, F.; Thom, J.T.; Hamann, A.; Frischbutter, S.; Baumgrass, R.; Chang, H.D.; Thiel, A.; Dong, J.; et al. NK cells gain higher IFN-γ competence during terminal differentiation. Eur. J. Immunol. 2014, 44, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Wiencke, J.K.; Butler, R.; Hsuang, G.; Eliot, M.; Kim, S.; Sepulveda, M.A.; Siegel, D.; Houseman, E.A.; Kelsey, K.T. The DNA methylation profile of activated human natural killer cells. Epigenetics 2016, 11, 363–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koues, O.I.; Collins, P.L.; Cella, M.; Robinette, M.L.; Porter, S.I.; Pyfrom, S.C.; Payton, J.E.; Colonna, M.; Oltz, E.M. Distinct Gene Regulatory Pathways for Human Innate versus Adaptive Lymphoid Cells. Cell 2016, 165, 1134–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.Y.; Sciumè, G.; Mikami, Y.; Guo, L.; Sun, H.W.; Brooks, S.R.; Urban, J.F.; Davis, F.P.; Kanno, Y.; O’Shea, J.J. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 2016, 165, 1120–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231.e13–1246.e13. [Google Scholar] [CrossRef] [PubMed]
- Tak, T.; Drylewicz, J.; Conemans, L.; De Boer, R.J.; Koenderman, L.; Borghans, J.A.M.; Tesselaar, K. Circulatory and maturation kinetics of human monocyte subsets in vivo. Blood 2017, 130, 1474–1477. [Google Scholar] [CrossRef] [Green Version]
- Coillard, A.; Segura, E. In vivo Differentiation of Human Monocytes. Front. Immunol. 2019, 10, 1907. [Google Scholar] [CrossRef]
- Mukherjee, R.; Kanti Barman, P.; Kumar Thatoi, P.; Tripathy, R.; Kumar Das, B.; Ravindran, B. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci. Rep. 2015, 5, 13886. [Google Scholar] [CrossRef] [Green Version]
- Gainaru, G.; Papadopoulos, A.; Tsangaris, I.; Lada, M.; Giamarellos-Bourboulis, E.J.; Pistiki, A. Increases in inflammatory and CD14dim/CD16pos/CD45pos patrolling monocytes in sepsis: Correlation with final outcome. Crit. Care 2018, 22, 56. [Google Scholar] [CrossRef] [Green Version]
- Puchner, A.; Saferding, V.; Bonelli, M.; Mikami, Y.; Hofmann, M.; Brunner, J.S.; Caldera, M.; Goncalves-Alves, E.; Binder, N.B.; Fischer, A.; et al. Non-classical monocytes as mediators of tissue destruction in arthritis. Ann. Rheum. Dis. 2018, 77, 1490–1497. [Google Scholar] [CrossRef] [Green Version]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Gomez Perdiguero, E.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M.I. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, K.F.; Neele, A.E.; Jukema, J.W.; Heijmans, B.T.; De Winther, M.P.J. Human monocyte-to-macrophage differentiation involves highly localized gain and loss of DNA methylation at transcription factor binding sites. Epigenetics Chromatin 2019, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gomez, A.; Li, T.; Kerick, M.; Català-Moll, F.; Comet, N.R.; Rodríguez-Ubreva, J.; De La Rica, L.; Branco, M.R.; Martín, J.; Ballestar, E. TET2- and TDG-mediated changes are required for the acquisition of distinct histone modifications in divergent terminal differentiation of myeloid cells. Nucleic Acids Res. 2017, 45, 10002–10017. [Google Scholar] [CrossRef]
- Li, T.; Garcia-Gomez, A.; Morante-Palacios, O.; Ciudad, L.; Özkaramehmet, S.; Van Dijck, E.; Rodríguez-Ubreva, J.; Vaquero, A.; Ballestar, E. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2019, 48, 665–681. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Gerrick, K.Y.; Gerrick, E.R.; Gupta, A.; Wheelan, S.J.; Yegnasubramanian, S.; Jaffee, E.M. Transcriptional profiling identifies novel regulators of macrophage polarization. PLoS ONE 2018, 13, e0208602. [Google Scholar] [CrossRef] [Green Version]
- Vento-Tormo, R.; Company, C.; Rodríguez-Ubreva, J.; de la Rica, L.; Urquiza, J.M.; Javierre, B.M.; Sabarinathan, R.; Luque, A.; Esteller, M.; Aran, J.M.; et al. IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. 2016, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ulm, A.; Somineni, H.K.; Oh, S.; Weirauch, M.T.; Zhang, H.-X.; Chen, X.; Lehn, M.A.; Janssen, E.M.; Ji, H. DNA methylation dynamics during ex vivo differentiation and maturation of human dendritic cells. Epigenetics Chromatin 2014, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacis, A.; Mailhot-Léonard, F.; Tailleux, L.; Randolph, H.E.; Yotova, V.; Dumaine, A.; Grenier, J.C.; Barreiro, L.B. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc. Natl. Acad. Sci. USA 2019, 116, 6938–6943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacis, A.; Tailleux, L.; Morin, A.M.; Lambourne, J.; MacIsaac, J.L.; Yotova, V.; Dumaine, A.; Danckært, A.; Luca, F.; Grenier, J.C.; et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015, 25, 1801–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; van der Meer, J.W.M. Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 2017, 21, 297–300. [Google Scholar] [CrossRef]
- de la Rica, L.; Rodríguez-Ubreva, J.; García, M.; Islam, A.B.M.M.K.; Urquiza, J.M.; Hernando, H.; Christensen, J.; Helin, K.; Gómez-Vaquero, C.; Ballestar, E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013, 14, R99. [Google Scholar] [CrossRef] [Green Version]
- Therapy, G.; Jin, S.-Y.; Xiao, J.; Bao, J.; Zhou, S.; Wright, J.F.; Long Zheng, X. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood 2013, 121, 3825–3829. [Google Scholar]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Zilbauer, M.; Rayner, T.F.; Clark, C.; Coffey, A.J.; Joyce, C.J.; Palta, P.; Palotie, A.; Lyons, P.A.; Smith, K.G.C. Genome-wide methylation analyses of primary human leukocyte subsets identifies functionally important cell-type-specific hypomethylated regions. Blood 2013, 122, e52–e60. [Google Scholar] [CrossRef] [Green Version]
- Ostuni, R.; Natoli, G.; Cassatella, M.A.; Tamassia, N. Epigenetic regulation of neutrophil development and function. Semin. Immunol. 2016, 28, 83–93. [Google Scholar] [CrossRef]
- Monticelli, S.; Leoni, C. Epigenetic and transcriptional control of mast cell responses. F1000Res. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Montagner, S.; Leoni, C.; Emming, S.; Della Chiara, G.; Balestrieri, C.; Barozzi, I.; Piccolo, V.; Togher, S.; Ko, M.; Rao, A.; et al. TET2 Regulates Mast Cell Differentiation and Proliferation through Catalytic and Non-catalytic Activities. Cell Rep. 2016, 15, 1566–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Levine, R.L.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.Y.; Pardanani, A.; et al. Frequent TET2 mutations in systemic mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 2009, 23, 900–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucie, E.; Hanssens, K.; Mercher, T.; Georgin-Lavialle, S.; Damaj, G.; Livideanu, C.; Chandesris, M.O.; Acin, Y.; Létard, S.; De Sepulveda, P.; et al. In aggressive forms of mastocytosis, TET2 loss cooperates with c-KITD816V to transform mast cells. Blood 2012, 120, 4846–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef] [Green Version]
- Grimbacher, B.; Warnatz, K.; Yong, P.F.K.; Korganow, A.S.; Peter, H.H. The crossroads of autoimmunity and immunodeficiency: Lessons from polygenic traits and monogenic defects. J. Allergy Clin. Immunol. 2016, 137, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef]
- Donlin, L.T.; Park, S.H.; Giannopoulou, E.; Ivovic, A.; Park-Min, K.H.; Siegel, R.M.; Ivashkiv, L.B. Insights into rheumatic diseases from next-generation sequencing. Nat. Rev. Rheumatol. 2019, 15, 327–339. [Google Scholar] [CrossRef]
- Plant, D.; Webster, A.; Nair, N.; Oliver, J.; Smith, S.L.; Eyre, S.; Hyrich, K.L.; Wilson, A.G.; Morgan, A.W.; Isaacs, J.D.; et al. Differential Methylation as a Biomarker of Response to Etanercept in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1353–1360. [Google Scholar] [CrossRef]
- Glossop, J.R.; Nixon, N.B.; Emes, R.D.; Sim, J.; Packham, J.C.; Mattey, D.L.; Farrell, W.E.; Fryer, A.A. DNA methylation at diagnosis is associated with response to disease-modifying drugs in early rheumatoid arthritis. Epigenomics 2017, 9, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Mok, A.; Rhead, B.; Holingue, C.; Shao, X.; Quach, H.L.; Quach, D.; Sinclair, E.; Graf, J.; Imboden, J.; Link, T.; et al. Hypomethylation of CYP2E1 and DUSP22 Promoters Associated With Disease Activity and Erosive Disease Among Rheumatoid Arthritis Patients. Arthritis Rheumatol. 2018, 70, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ubreva, J.; De La Calle-Fabregat, C.; Li, T.; Ciudad, L.; Ballestar, M.L.; Català-Moll, F.; Morante-Palacios, O.; Garcia-Gomez, A.; Celis, R.; Humby, F.; et al. Inflammatory cytokines shape a changing DNA methylome in monocytes mirroring disease activity in rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, J.W.; Shoemaker, R.; Boyle, D.L.; Hillman, J.; Anderson, D.; Wang, W.; Firestein, G.S. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med. 2013, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Ai, R.; Laragione, T.; Hammaker, D.; Boyle, D.L.; Wildberg, A.; Maeshima, K.; Palescandolo, E.; Krishna, V.; Pocalyko, D.; Whitaker, J.W.; et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 2018, 9, 1921. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928. [Google Scholar] [CrossRef]
- Imgenberg-Kreuz, J.; Almlöf, J.C.; Leonard, D.; Alexsson, A.; Nordmark, G.; Eloranta, M.L.; Rantapää-Dahlqvist, S.; Bengtsson, A.A.; Jönsen, A.; Padyukov, L.; et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Lanata, C.M.; Paranjpe, I.; Nititham, J.; Taylor, K.E.; Gianfrancesco, M.; Paranjpe, M.; Andrews, S.; Chung, S.A.; Rhead, B.; Barcellos, L.F.; et al. A phenotypic and genomics approach in a multi-ethnic cohort to subtype systemic lupus erythematosus. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility andtranscriptional poising of interferon-regulated genes in naïve CD4+ T cellsfrom lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Wang, J.; Liao, W.; Li, D.; Li, M.; Wu, H.; Zhang, Y.; Gershwin, M.E.; Lu, Q. Increased 5-hydroxymethylcytosine in CD4+ T cells in systemic lupus erythematosus. J. Autoimmun. 2016, 69, 64–73. [Google Scholar] [CrossRef]
- Absher, D.M.; Li, X.; Waite, L.L.; Gibson, A.; Roberts, K.; Edberg, J.; Chatham, W.W.; Kimberly, R.P. Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations. PLoS Genet. 2013, 9, 1003678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulff-Møller, C.J.; Asmar, F.; Liu, Y.; Svendsen, A.J.; Busato, F.; Grønbæk, K.; Tost, J.; Jacobsen, S. Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 878–890. [Google Scholar] [CrossRef] [Green Version]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [Green Version]
- Aubert, P.; Suárez-Fariñas, M.; Mitsui, H.; Johnson-Huang, L.M.; Harden, J.L.; Pierson, K.C.; Dolan, J.G.; Novitskaya, I.; Coats, I.; Estes, J.; et al. Homeostatic Tissue Responses in Skin Biopsies from NOMID Patients with Constitutive Overproduction of IL-1β. PLoS ONE 2012, 7, e49408. [Google Scholar] [CrossRef] [PubMed]
- Erdem, G.C.; Erdemir, S.; Abaci, I.; Aydin, A.K.K.; Everest, E.; Turanli, E.T. Alternatively spliced MEFV transcript lacking exon 2 and its protein isoform pyrin-2d implies an epigenetic regulation of the gene in inflammatory cell culture models. Genet. Mol. Biol. 2017, 40, 688–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirectepe, A.K.; Erdem, G.C.; Senturk, N.; Arisoy, N.; Hatemi, G.; Ozdogan, H.; Kasapcopur, O.; Tahir Turanli, E. Increased expression of exon 2 deleted MEFV transcript in Familial Mediterranean Fever patients. Int. J. Immunogenet. 2011, 38, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Kamae, C.; Imai, K.; Kato, T.; Okano, T.; Honma, K.; Nakagawa, N.; Yeh, T.-W.; Noguchi, E.; Ohara, A.; Shigemura, T.; et al. Clinical and Immunological Characterization of ICF Syndrome in Japan. J. Clin. Immunol. 2018, 38, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Walton, E.L.; Sterlin, D.; Hédouin, S.; Nitta, H.; Ito, Y.; Fouyssac, F.; Mégarbané, A.; Sasaki, H.; Picard, C.; et al. Germline genes hypomethylation and expression define a molecular signature in peripheral blood of ICF patients: Implications for diagnosis and etiology. Orphanet J. Rare Dis. 2014, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.R.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [Green Version]
- Picard, C.; Puel, A.; Bonnet, M.; Ku, C.-L.; Bustamante, J.; Yang, K.; Soudais, C.; Dupuis, S.; Feinberg, J.; Fieschi, C.; et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003, 299, 2076–2079. [Google Scholar] [CrossRef]
- Davidson, D.J.; Currie, A.J.; Bowdish, D.M.E.; Brown, K.L.; Rosenberger, C.M.; Ma, R.C.; Bylund, J.; Campsall, P.A.; Puel, A.; Picard, C.; et al. IRAK-4 Mutation (Q293X): Rapid Detection and Characterization of Defective Post-Transcriptional TLR/IL-1R Responses in Human Myeloid and Non-Myeloid Cells. J. Immunol. 2006, 177, 8202–8211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, S.; Thomson, B.P.; Remmers, E.F.; Eyre, S.; Hinks, A.; Guiducci, C.; Catanese, J.J.; Xie, G.; Stahl, E.A.; Chen, R.; et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat. Genet. 2009, 41, 1313–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Lanata, C.M.; Chung, S.A.; Criswell, L.A. DNA methylation 101: What is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci. Med. 2018, 5, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birney, E.; Smith, G.D.; Greally, J.M. Epigenome-wide Association Studies and the Interpretation of Disease -Omics. PLoS Genet. 2016, 12, e1006105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Ignacio Martin-Subero, J.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Keddie, S.; Parker, T.; Lachmann, H.J.; Ginsberg, L. Cryopyrin-Associated Periodic Fever Syndrome and the Nervous System. Curr. Treat. Opt. Neurol. 2018, 20, 43. [Google Scholar] [CrossRef] [Green Version]
- Aksentijevich, I.; Putnam, C.D.; Remmers, E.F.; Mueller, J.L.; Le, J.; Kolodner, R.D.; Moak, Z.; Chuang, M.; Austin, F.; Goldbach-Mansky, R.; et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007, 56, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Vento-Tormo, R.; Álvarez-Errico, D.; Garcia-Gomez, A.; Hernández-Rodríguez, J.; Buján, S.; Basagaña, M.; Méndez, M.; Yagüe, J.; Juan, M.; Aróstegui, J.I.; et al. DNA demethylation of inflammasome-associated genes is enhanced in patients with cryopyrin-associated periodic syndromes. J. Allergy Clin. Immunol. 2017, 139, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Komarow, H.D.; Cheng, J.; Wood, G.; Raben, N.; Liu, P.P.; Kastner, D.L. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 2003, 11, 591–604. [Google Scholar] [CrossRef]
- Yu, J.W.; Wu, J.; Zhang, Z.; Datta, P.; Ibrahimi, I.; Taniguchi, S.; Sagara, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Cryopyrin and pyrin activate caspase-1, but not NF-κB, via ASC oligomerization. Cell Death Differ. 2006, 13, 236–249. [Google Scholar] [CrossRef] [Green Version]
- Seshadri, S.; Duncan, M.D.; Hart, J.M.; Gavrilin, M.A.; Wewers, M.D. Pyrin Levels in Human Monocytes and Monocyte-Derived Macrophages Regulate IL-1β Processing and Release. J. Immunol. 2007, 179, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, M.A.; Abdelaziz, D.H.A.; Mostafa, M.; Abdulrahman, B.A.; Grandhi, J.; Akhter, A.; Abu Khweek, A.; Aubert, D.F.; Valvano, M.A.; Wewers, M.D.; et al. Activation of the Pyrin Inflammasome by Intracellular Burkholderia cenocepacia. J. Immunol. 2012, 188, 3469–3477. [Google Scholar] [CrossRef] [Green Version]
- Chae, J.J.; Cho, Y.H.; Lee, G.S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity 2011, 34, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Sohar, E.; Gafni, J.; Pras, M.; Heller, H. Familial Mediterranean Fever. A survey of 470 cases and review of the literature. Am. J. Med. 1967, 43, 227–253. [Google Scholar] [CrossRef]
- Booty, M.G.; Jae, J.C.; Masters, S.L.; Remmers, E.F.; Barham, B.; Le, J.M.; Barron, K.S.; Holland, S.M.; Kastner, D.L.; Aksentijevich, I. Familial Mediterranean Fever with a single MEFV mutation: Where is the second hit? Arthritis Rheum. 2009, 60, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Rowczenio, D.M.; Gomes, S.M.; Aróstegui, J.I.; Mensa-Vilaro, A.; Omoyinmi, E.; Trojer, H.; Baginska, A.; Baroja-Mazo, A.; Pelegrin, P.; Savic, S.; et al. Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP3 mosaicism-UK single center experience. Front. Immunol. 2017, 8, 1410. [Google Scholar] [CrossRef]
- Marek-Yagel, D.; Berkun, Y.; Padeh, S.; Abu, A.; Reznik-Wolf, H.; Livneh, A.; Pras, M.; Pras, E. Clinical disease among patients heterozygous for Familial Mediterranean Fever. Arthritis Rheum. 2009, 60, 1862–1866. [Google Scholar] [CrossRef]
- Kirectepe, A.K.; Kasapcopur, O.; Arisoy, N.; Celikyapi Erdem, G.; Hatemi, G.; Ozdogan, H.; Tahir Turanli, E. Analysis of MEFV exon methylation and expression patterns in Familial Mediterranean Fever. BMC Med. Genet. 2011, 12, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulum, Y.A.; Peynircioglu, B.B.; Batu, E.; Guler, C.; Karadag, O.; Ertenli, A.; Kiraz, S.; Ozen, S.; Yilmaz, E. MEFV gene methylation pattern analysis in Familial Mediterranean Fever patients with altered expression levels. Pediatr. Rheumatol. 2015, 13, P113. [Google Scholar] [CrossRef] [Green Version]
- Simo-Riudalbas, L.; Diaz-Lagares, A.; Gatto, S.; Gagliardi, M.; Crujeiras, A.B.; Matarazzo, M.R.; Esteller, M.; Sandoval, J. Genome-Wide DNA methylation analysis identifies novel hypomethylated non- Pericentromeric genes with potential clinical implications in ICF syndrome. PLoS ONE 2015, 10, e0132517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, M.E.; Lana, E.; Rivals, I.; Lefranc, G.; Sarda, P.; Claustres, M.; Mégarbané, A.; de Sario, A. Heterochromatic genes undergo epigenetic changes and escape silencing in immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome. PLoS ONE 2011, 6, e19464. [Google Scholar] [CrossRef]
- Ehrlich, M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin. Immunol. 2003, 109, 17–28. [Google Scholar] [CrossRef]
- Pezzolo, A.; Prigione, I.; Facchetti, P.; Castellano, E.; Viale, M.; Gimelli, G.; Pistoia, V. T-cell apoptosis in ICF syndrome. J. Allergy Clin. Immunol. 2001, 108, 310–312. [Google Scholar] [CrossRef]
- Rechavi, E.; Lev, A.; Eyal, E.; Barel, O.; Kol, N.; Barhom, S.F.; Pode-Shakked, B.; Anikster, Y.; Somech, R.; Simon, A.J. A Novel Mutation in a Critical Region for the Methyl Donor Binding in DNMT3B Causes Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome (ICF). J. Clin. Immunol. 2016, 36, 801–809. [Google Scholar] [CrossRef]
- Hernandez, R.; Frady, A.; Zhang, X.Y.; Varela, M.; Ehrlich, M. Preferential induction of chromosome 1 multibranched figures and whole-arm deletions in a human pro-B cell line treated with 5-azacytidine or 5-azadeoxycytidine. Cytogenet. Genome Res. 1997, 76, 196–201. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J. Biol. Chem. 2002, 277, 20409–20414. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Okano, M.; Williams, C.; Chen, T.; Georgopoulos, K.; Li, E. Roles for Dnmt3b in mammalian development: A mouse model for the ICF syndrome. Development 2006, 133, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyn, H.; Vida, E.; Sayols, S.; Sanchez-Mut, J.V.; Moran, S.; Medina, I.; Sandoval, J.; Simó-Riudalbas, L.; Szczesna, K.; Huertas, D.; et al. Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient. Epigenetics 2012, 7, 542–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, B.; Tao, Q.; Peng, J.; Soo, H.M.; Wu, W.; Ying, J.; Fields, C.R.; Delmas, A.I.; Liu, X.; Qiu, J.; et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008, 17, 690–709. [Google Scholar] [CrossRef] [Green Version]
- De Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, P.E.; Ito, Y.; Grillo, G.; Wang, J.; Velasco, G.; Nitta, H.; Unoki, M.; Yoshihara, M.; Suyama, M.; Sun, Y.; et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015, 6, 7870. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.J.; Kaur, R.; Sosa, C.P.; Lee, J.H.; Kashiwagi, K.; Zhou, D.; Robertson, K.D. ZBTB24 is a transcriptional regulator that coordinates with DNMT3B to control DNA methylation. Nucleic Acids Res. 2018, 46, 10034–10051. [Google Scholar] [CrossRef]
- Ren, R.; Hardikar, S.; Horton, J.R.; Lu, Y.; Zeng, Y.; Singh, A.K.; Lin, K.; Coletta, L.D.; Shen, J.; Lin Kong, C.S.; et al. Structural basis of specific DNA binding by the transcription factor ZBTB24. Nucleic Acids Res. 2019, 47, 8388–8398. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Geiman, T.M.; Xi, S.; Jiang, Q.; Schmidtmann, A.; Chen, T.; Li, E.; Muegge, K. Lsh is involved in de novo methylation of DNA. EMBO J. 2006, 25, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Briones, V.; Barbour, S.; Yu, W.; Han, Y.; Terashima, M.; Muegge, K. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015, 43, 1444–1455. [Google Scholar] [CrossRef]
- Yu, W.; McIntosh, C.; Lister, R.; Zhu, I.; Han, Y.; Ren, J.; Landsman, D.; Lee, E.; Briones, V.; Terashima, M.; et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014, 24, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Thijssen, P.E.; de Klerk, E.; Vonk, K.K.D.; Wang, J.; den Hamer, B.; Aytekin, C.; van der Maarel, S.M.; Daxinger, L. Converging disease genes in ICF syndrome: ZBTB24 controls expression of CDCA7 in mammals. Hum. Mol. Genet. 2016, 25, 4041–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenness, C.; Giunta, S.; Müller, M.M.; Kimura, H.; Muir, T.W.; Funabiki, H. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E876–E885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, G.; Grillo, G.; Touleimat, N.; Ferry, L.; Ivkovic, I.; Ribierre, F.; Deleuze, J.F.; Chantalat, S.; Picard, C.; Francastel, C. Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum. Mol. Genet. 2018, 27, 2409–2424. [Google Scholar] [CrossRef] [Green Version]
- Weemaes, C.M.; Van Tol, M.J.; Wang, J.; Van Ostaijen-Ten Dam, M.M.; Van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; Van Der Burg, M.; Graham Davies, E.; et al. Heterogeneous clinical presentation in icf syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Barlan, I.; Chapel, H.; Costa-Carvalho, B.T.; Cunningham-Rundles, C.; de la Morena, M.T.; Espinosa-Rosales, F.J.; Hammarström, L.; Nonoyama, S.; Quinti, I.; et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract. 2016, 4, 38–59. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Cortez, V.C.; Del Pino-Molina, L.; Rodríguez-Ubreva, J.; Ciudad, L.; Gómez-Cabrero, D.; Company, C.; Urquiza, J.M.; Tegnér, J.; Rodríguez-Gallego, C.; López-Granados, E.; et al. Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naïve-to-memory B-cell transition. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Del Pino-Molina, L.; Rodríguez-Ubreva, J.; Canizales, J.T.; Coronel-Díaz, M.; Kulis, M.; Martín-Subero, J.I.; Van Der Burg, M.; Ballestar, E.; López-Granados, E. Impaired CpG demethylation in common variable immunodeficiency associates with B cell phenotype and proliferation rate. Front. Immunol. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Von Bernuth, H.; Picard, C.; Jin, Z.; Pankla, R.; Xiao, H.; Ku, C.-L.; Chrabieh, M.; Mustapha, I.B.; Ghandil, P.; Camcioglu, Y.; et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 2008, 321, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y.; Herman, M.; Ciancanelli, M.J.; Pérez de Diego, R.; Sancho-Shimizu, V.; Abel, L.; Casanova, J.L. TLR3 immunity to infection in mice and humans. Curr. Opin. Immunol. 2013, 25, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.; Yeung, J.; Hildebrand, K.J.; Junker, A.K.; Turvey, S.E. Human primary immunodeficiencies causing defects in innate immunity. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 607–613. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Immune Activity | Response Affected | Disease | Monogenic/Complex | Etiology | Cell types with Known DNA Methylation Defects |
|---|---|---|---|---|---|
| Overactivation | Adaptive | RA | Polygenic (20–50% heritability). HLA association: DRB1*01 and *04. Non-HLA SNP association: PTPN22, IL23R, TRAF1, CTLA4 | Mostly unknown. Disease risk factors: age, cigarette smoke | Whole blood [100], PBLs [98], T cells [101,102], Monocytes [102,103]; B-cells [102], Fibroblast-like synoviocytes [104,105,106,107] |
| SLE | Polygenic (4–20% heritability). HLA association: DRB1*1501 and DRB1*0301. Non-HLA SNP association: IRFs, STAT4, IFIH1, OPN | Mostly unknown. Disease risk factors: ultraviolet light, cigarette smoke | Whole Blood [108,109,110], T cells [111,112], B cells [113,114], Monocytes118 | ||
| Innate | CAPS | Monogenic | NRLP3 GoF mutations | Monocytes [115] | |
| FMF | Monogenic | MEFV GoF mutations | PBMCs [116,117] | ||
| Defect | Adaptive | ICF | Monogenic | DNMT3B LoF mutations | Fibroblasts [118], Lymphoblastoid Cell Line [118,119,120] |
| CVID | Polygenic (5–25% heritability) | Mostly unknown. Patients may present SNPs/mutations in CTLA4, LRBA, TNFRSF13B, MSH5 | B Cells [121,122] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calle-Fabregat, C.d.l.; Morante-Palacios, O.; Ballestar, E. Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease. Genes 2020, 11, 110. https://doi.org/10.3390/genes11010110
Calle-Fabregat Cdl, Morante-Palacios O, Ballestar E. Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease. Genes. 2020; 11(1):110. https://doi.org/10.3390/genes11010110
Chicago/Turabian StyleCalle-Fabregat, Carlos de la, Octavio Morante-Palacios, and Esteban Ballestar. 2020. "Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease" Genes 11, no. 1: 110. https://doi.org/10.3390/genes11010110
APA StyleCalle-Fabregat, C. d. l., Morante-Palacios, O., & Ballestar, E. (2020). Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease. Genes, 11(1), 110. https://doi.org/10.3390/genes11010110