Does DNA Methylation Matter in FSHD?



Abstract
:1. Introduction
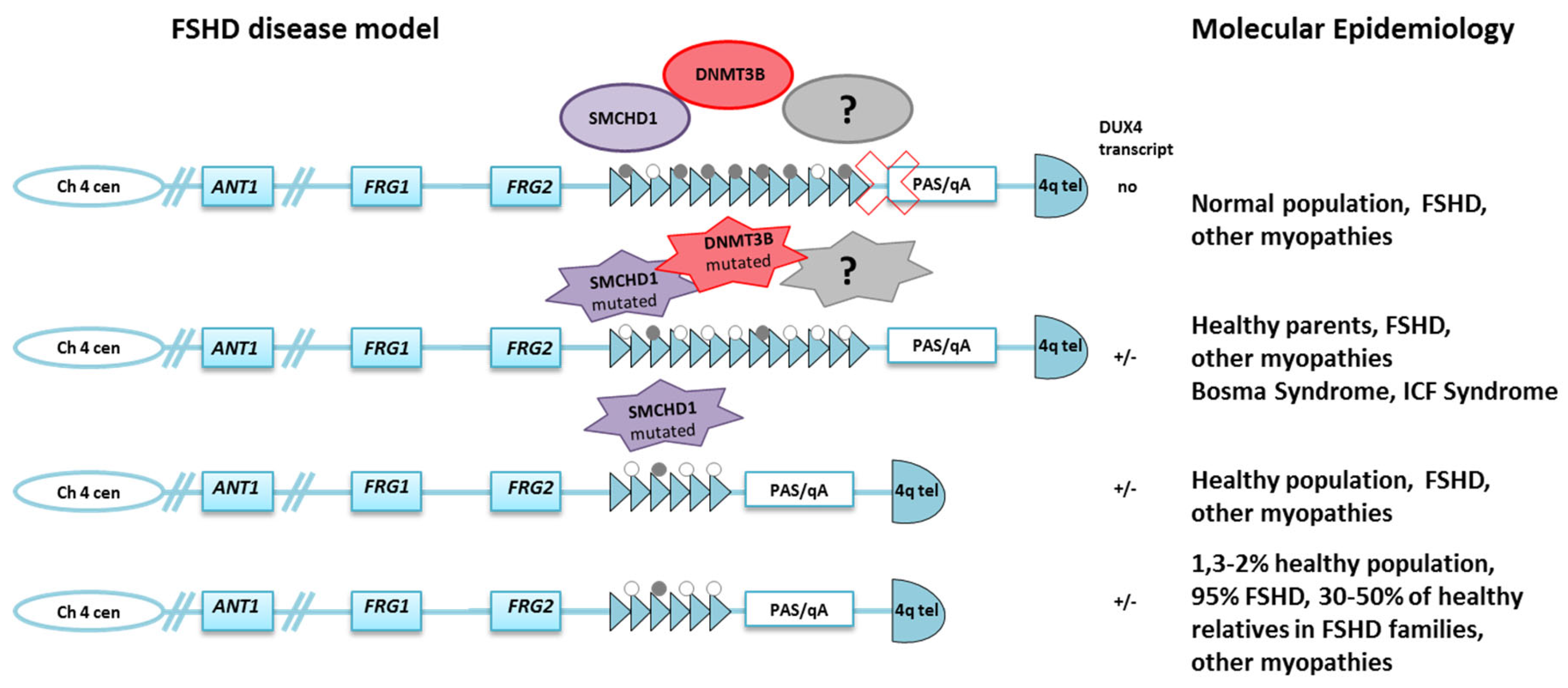
2. Molecular Features and the Epigenetic Model for FSHD
3. The Debated Role of DNA Methylation in FSHD: Clinical and Families Studies
4. DNA Methylation in FSHD2
5. Changes in DNA Methylation at D4Z4 Upon Reprogramming
6. Trans-Acting Factors
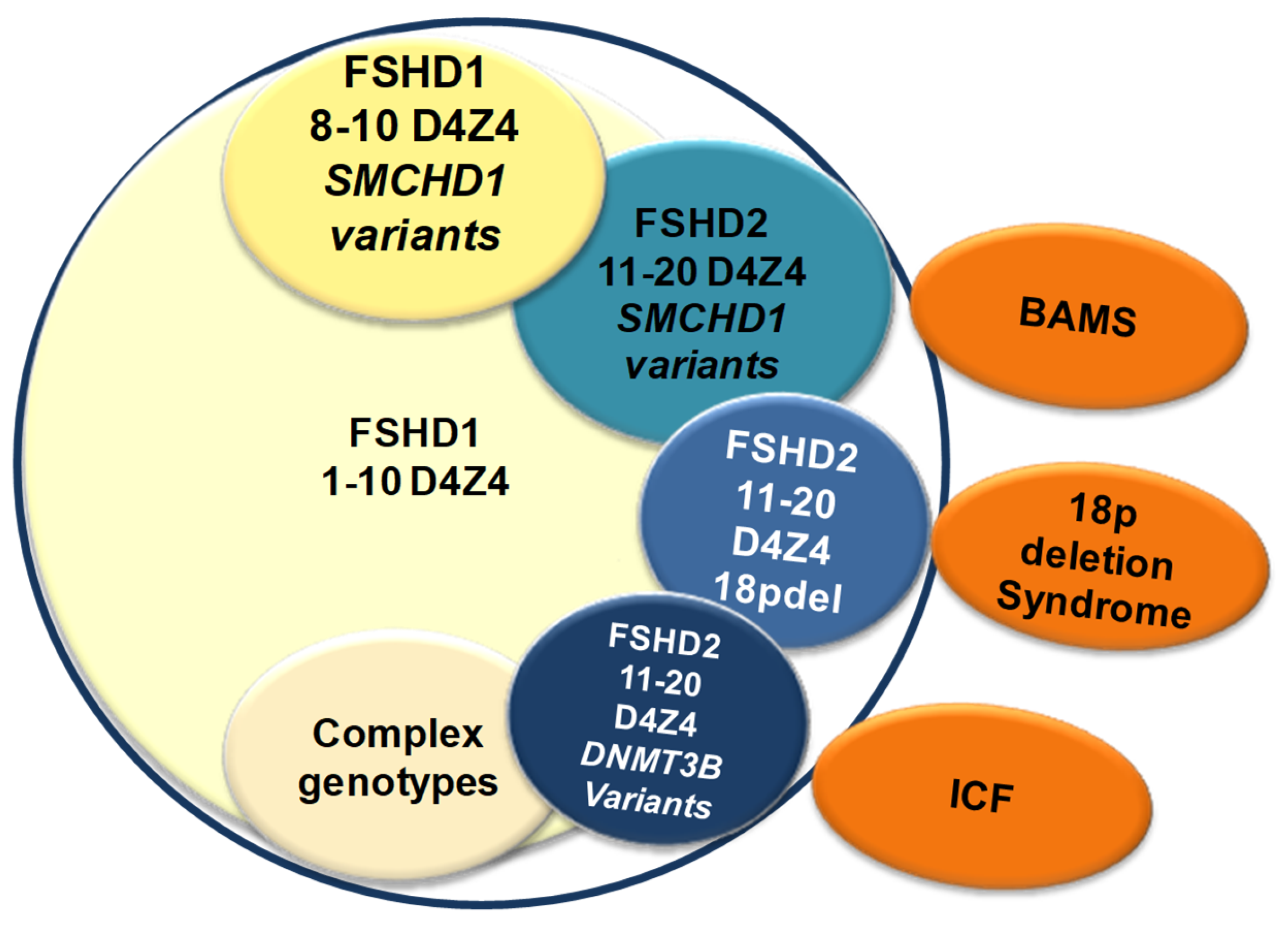
7. The Significance of D4Z4 Hypomethylation in FSHD and Its Implication for Clinical Counseling
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deenen, J.C.W.; Arnts, H.; Van Der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.M.; Van Engelen, B.G.M. EPopulation-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostacciuolo, M.L.; Pastorello, E.; Vazza, G.; Miorin, M.; Angelini, C.; Tomelleri, G.; Galluzzi, G.; Trevisan, C.P. Facioscapulohumeral muscular dystrophy: Epidemiological and molecular study in a north-east Italian population sample. Clin. Genet. 2009, 75, 550–555. [Google Scholar] [CrossRef]
- Padberg, G.W.A.M.; LUMC. Facioscapulohumeral Disease. Ph.D. Thesis, Leiden University, Leiden, The Netherlands, 1982. [Google Scholar]
- Flanigan, K.M. Facioscapulohumeral Muscular Dystrophy and Scapulohumeral Syndrome; McGraw-Hill: New York, NY, USA, 2004. [Google Scholar]
- Zatz, M.; Marie, S.K.; Passos-Bueno, M.R.; Vainzof, M.; Campiotto, S.; Cerqueira, A.; Wijmenga, C.; Padberg, G.; Frants, R. High proportion of new mutations and possible anticipation in Brazilian facioscapulohumeral muscular dystrophy families. Am. J. Hum. Genet. 1995, 56, 99–105. [Google Scholar] [PubMed]
- Tonini, M.M.O.; Passos-Bueno, M.R.; Cerqueira, A.; Matioli, S.R.; Pavanello, R.; Zatz, M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 2004, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; Scionti, I.; Sera, F.; Govi, M.; D’Amico, R.; Frambolli, I.; Mele, F.; Filosto, M.; Vercelli, L.; Ruggiero, L.; et al. Large scale genotype–phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 2013, 136, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Hong, J.M.; Lee, J.H.; Lee, H.S.; Shin, H.Y.; Kim, S.M.; Ki, C.S.; Lee, J.H.; Choi, Y.C. Low D4Z4 copy number and gender difference in Korean patients with facioscapulohumeral muscular dystrophy type 1. Neuromuscul. Disord. 2015, 25, 859–864. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Wang, Z.Q.; Murong, S.X.; Wang, N. FSHD in Chinese population: Characteristics of translocation and genotype-phenotype correlation. Neurology 2004, 63, 581–583. [Google Scholar] [CrossRef]
- Lin, F.; Wang, Z.-Q.; Lin, M.-T.; Murong, S.-X.; Wang, N. New Insights into Genotype-phenotype Correlations in Chinese Facioscapulohumeral Muscular Dystrophy. Chin. Med. J. (Engl). 2015, 128, 1707–1713. [Google Scholar] [CrossRef]
- Tawil, R.; Kissel, J.T.; Heatwole, C.; Pandya, S.; Gronseth, G.; Benatar, M.; Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology; Practice Issues Review Panel of the American Association of Neuromuscular Electrodiagnostic Medicine. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy. Neurology 2015, 85, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Goto, K.; Nishino, I.; Hayashi, Y.K. Very low penetrance in 85 Japanese families with facioscapulohumeral muscular dystrophy 1A. J. Med. Genet. 2004, 41, e12. [Google Scholar] [CrossRef] [Green Version]
- Tawil, R.; Storvick, D.; Feasby, T.E.; Weiffenbach, B.; Griggs, R.C. Extreme variability of expression in monozygotic twins with FSH muscular dystrophy. Neurology 1993, 43, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Tawil, R.; McDermott, M.; Forrester, J.; Figlewicz, D.; Weiffenbach, B. Monozygotic twins with facioscapulohumeral dystrophy (FSHD): Implications for genotype/phenotype correlation. Muscle Nerve 1995, 18, S50–S55. [Google Scholar] [CrossRef]
- Tupler, R.; Barbierato, L.; Memmi, M.; Sewry, C.A.; De Grandis, D.; Maraschio, P.; Tiepolo, L.; Ferlini, A. Identical de novo mutation at the D4F104S1 locus in monozygotic male twins affected by facioscapulohumeral muscular dystrophy (FSHD) with different clinical expression. J. Med. Genet. 1998, 35, 778–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijmenga, C.; Frants, R.R.; Brouwer, O.F.; van der Klift, H.M.; Khan, P.M.; Padberg, G.W. Facioscapulohumeral muscular dystrophy gene in Dutch families is not linked to markers for familial adenomatous polyposis on the long arm of chromosome 5. J. Neurol. Sci. 1990, 95, 225–229. [Google Scholar] [CrossRef]
- Padberg, G.W.; Lunt, P.W.; Koch, M.; Fardeau, M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991, 1, 231–234. [Google Scholar] [CrossRef]
- Weiffenbach, B.; Bagley, R.G.; Falls, K.; Dubois, J.; Hyser, C.; Storvick, D.; Schultz, P.; Mendell, J.R.; Milner, E.C.; Jacobsen, S.J. Framework multipoint map of the long arm of human chromosome 4 and telomeric localization of the gene for FSHD. Mamm. Genome 1992, 3, 143–150. [Google Scholar] [CrossRef]
- Sarfarazi, M.; Wijmenga, C.; Upadhyaya, M.; Weiffenbach, B.; Hyser, C.; Mathews, K.; Murray, J.; Gilbert, J.; Pericak-Vance, M.; Lunt, P. Regional mapping of facioscapulohumeral muscular dystrophy gene on 4q35: Combined analysis of an international consortium. Am. J. Hum. Genet. 1992, 51, 396–403. [Google Scholar]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.-M.; Hofker, M.H.; Moerer, P.; Williamson, R.; et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef]
- Tremblay, D.C.; Alexander, G.; Moseley, S.; Chadwick, B.P. Expression, tandem repeat copy number variation and stability of four macrosatellite arrays in the human genome. BMC Genom. 2010, 11, 632. [Google Scholar] [CrossRef] [Green Version]
- Lunt, P.W.; Jardine, P.E.; Koch, M.; Maynard, J.; Osborn, M.; Williams, M.; Harper, P.S.; Upadhyaya, M. Phenotypic-genotypic correlation will assist genetic counseling in 4q35-facioscapulohumeral muscular dystrophy. Muscle Nerve. Suppl. 1995, 2, S103–S109. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Maynard, J.; Rogers, M.T.; Lunt, P.W.; Jardine, P.; Ravine, D.; Harper, P.S. Improved molecular diagnosis of facioscapulohumeral muscular dystrophy (FSHD): Validation of the differential double digestion for FSHD. J. Med. Genet. 1997, 34, 476–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawil, R.; Van Der Maarel, S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- NAKAGAWA, M.; MATSUZAKI, T.; HIGUCHI, I.; FUKUNAGA, H.; INUI, T.; NAGAMITSU, S.; YAMADA, H.; ARIMURA, K.; OSAME, M. Facioscapulohumeral Muscular Dystrophy: Clinical Diversity and Genetic Abnormalities in Japanese Patients. Intern. Med. 1997, 36, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlgemuth, M.; Lemmers, R.J.; van der Kooi, E.L.; van der Wielen, M.J.; van Overveld, P.G.; Dauwerse, H.; Bakker, E.; Frants, R.R.; Padberg, G.W.; van der Maarel, S.M. Possible phenotypic dosage effect in patients compound heterozygous for FSHD-sized 4q35 alleles. Neurology 2003, 61, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Scionti, I.; Greco, F.; Ricci, G.; Govi, M.; Arashiro, P.; Vercelli, L.; Berardinelli, A.; Angelini, C.; Antonini, G.; Cao, M.; et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2012, 90, 628–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overveld, P.G.M.v.; Lemmers, R.J.; Deidda, G.; Sandkuijl, L.; Padberg, G.W.; Frants, R.R.; van der Maarel, S.M. Interchromosomal repeat array interactions between chromosomes 4 and 10: A model for subtelomeric plasticity. Hum. Mol. Genet. 2000, 9, 2879–2884. [Google Scholar] [CrossRef] [Green Version]
- Ricci, G.; Ruggiero, L.; Vercelli, L.; Sera, F.; Nikolic, A.; Govi, M.; Mele, F.; Daolio, J.; Angelini, C.; Antonini, G.; et al. A novel clinical tool to classify facioscapulohumeral muscular dystrophy phenotypes. J. Neurol. 2016, 263, 1204–1214. [Google Scholar] [CrossRef]
- Ricci, G.; Zatz, M.; Tupler, R. Facioscapulohumeral Muscular Dystrophy: More Complex than it Appears. Curr. Mol. Med. 2014, 14, 1052–1068. [Google Scholar] [CrossRef]
- Butz, M.; Koch, M.C.; Muller-Felber, W.; Lemmers, R.J.L.F.; van der Maarel, S.M.; Schreiber, H. Facioscapulohumeral muscular dystrophy. J. Neurol. 2003, 250, 932–937. [Google Scholar] [CrossRef]
- de Greef, J.C.; Lemmers, R.J.L.F.; Camano, P.; Day, J.W.; Sacconi, S.; Dunand, M.; van Engelen, B.G.M.; Kiuru-Enari, S.; Padberg, G.W.; Rosa, A.L.; et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010, 75, 1548–1554. [Google Scholar] [CrossRef] [Green Version]
- van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Salort-Campana, E.; Nguyen, K.; Bernard, R.; Jouve, E.; Solé, G.; Nadaj-Pakleza, A.; Niederhauser, J.; Charles, E.; Ollagnon, E.; Bouhour, F.; et al. Low penetrance in facioscapulohumeral muscular dystrophy type 1 with large pathological D4Z4 alleles: A cross-sectional multicenter study. Orphanet J. Rare Dis. 2015, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, P.; Kekou, K.; Fryssira, H.; Sofocleous, C.; Manta, P.; Panousopoulou, A.; Gounaris, K.; Kanavakis, E. Mutation spectrum and phenotypic manifestation in FSHD Greek patients. Neuromuscul. Disord. 2012, 22, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.E.; Lyle, R.; Clark, L.N.; Valleley, E.M.; Wright, T.J.; Wijmenga, C.; van Deutekom, J.C.T.; Francis, F.; Sharpe, P.T.; Hofker, M.; et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystropothhy. Hum. Mol. Genet. 1994, 3, 1287–1295. [Google Scholar] [CrossRef]
- Bakker, E.; Wijmenga, C.; Vossen, R.H.; Padberg, G.W.; Hewitt, J.; van der Wielen, M.; Rasmussen, K.; Frants, R.R. The FSHD-linked locus D4F104S1 (p13E-11) on 4q35 has a homologue on 10qter. Muscle Nerve. Suppl. 1995, 2, S39–S44. [Google Scholar] [CrossRef] [Green Version]
- Deidda, G.; Cacurri, S.; Grisanti, P.; Vigneti, E.; Piazzo, N.; Felicetti, L. Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur. J. Hum. Genet. 1995, 3, 155–167. [Google Scholar] [CrossRef]
- Gabellini, D.; Green, M.R.; Tupler, R. Inappropriate gene activation in FSHD: A repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 2002, 110, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-M.; Yao, Y.L.; Seto, E. The FK506-binding protein 25 functionally associates with histone deacetylases and with transcription factor YY1. EMBO J. 2001, 20, 4814–4825. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol. 2001, 21, 5979–5991. [Google Scholar] [CrossRef] [Green Version]
- Oei, S.L.; Shi, Y. Transcription Factor Yin Yang 1 Stimulates Poly(ADP-Ribosyl)ation and DNA Repair. Biochem. Biophys. Res. Commun. 2001, 284, 450–454. [Google Scholar] [CrossRef]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012, 149, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massah, S.; Hollebakken, R.; Labrecque, M.P.; Kolybaba, A.M.; Beischlag, T.V.; Prefontaine, G.G. Epigenetic characterization of the growth hormone gene identifies SmcHD1 as a regulator of autosomal gene clusters. PLoS ONE 2014, 9, e97535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Hu, J.; Moore, D.L.; Liu, R.; Kessans, S.A.; Breslin, K.; Lucet, I.S.; Keniry, A.; Leong, H.S.; Parish, C.L.; et al. Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl. Acad. Sci. USA 2015, 112, E3535–E3544. [Google Scholar] [CrossRef] [Green Version]
- Mould, A.W.; Pang, Z.; Pakusch, M.; Tonks, I.D.; Stark, M.; Carrie, D.; Mukhopadhyay, P.; Seidel, A.; Ellis, J.J.; Deakin, J.; et al. Smchd1 regulates a subset of autosomal genes subject to monoallelic expression in addition to being critical for X inactivation. Epigenetics Chromatin 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Boogaard, M.L.; Lemmers, R.J.L.F.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; Van Der Vliet, P.J.; Straasheijm, K.R.; Van Den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Greef, J.C.; Lemmers, R.J.L.F.; van Engelen, B.G.M.; Sacconi, S.; Venance, S.L.; Frants, R.R.; Tawil, R.; van der Maarel, S.M. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum. Mutat. 2009, 30, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- de Greef, J.C.; Wohlgemuth, M.; Chan, O.A.; Hansson, K.B.; Smeets, D.; Frants, R.R.; Weemaes, C.M.; Padberg, G.W.; van der Maarel, S.M. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 2007, 69, 1018–1026. [Google Scholar] [CrossRef]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Mattéotti, C.; Van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [Green Version]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Mattéotti, C.; Arias, C.; Corona, E.D.; Nuñez, N.G.; Leo, O.; et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.L.F.; Van Der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; Van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; Mccune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Horard, B.; Eymery, A.; Fourel, G.; Vassetzky, N.; Puechberty, J.; Roizes, G.; Lebrigand, K.; Barbry, P.; Laugraud, A.; Gautier, C.; et al. Global analysis of DNA methylation and transcription of human repetitive sequences. Epigenetics 2009, 4, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Francastel, C.; Magdinier, F. DNA methylation in satellite repeats disorders. Essays Biochem. 2019, 63, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Cantone, I.; Fisher, A.G. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol. 2013, 20, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Toyota, M.; Issa, J.P.J. The role of DNA hypermethylation in human neoplasia. Electrophoresis 2000, 21, 329–333. [Google Scholar] [CrossRef]
- Bocker, M.T.; Hellwig, I.; Breiling, A.; Eckstein, V.; Ho, A.D.; Lyko, F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 2011, 117, e182–e189. [Google Scholar] [CrossRef] [Green Version]
- Martino, D.J.; Tulic, M.K.; Gordon, L.; Hodder, M.; Richman, T.; Metcalfe, J.; Prescott, S.L.; Saffery, R. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics 2011, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Feinberg, A.P. A genetic approach to cancer epigenetics. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta 2007, 1775, 138–162. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Mossevelde, S.; Van Der Zee, J.; Sieben, A.; Engelborghs, S.; De Bleecker, J.; Ivanoiu, A.; Deryck, O.; Edbauer, D.; Zhang, M.; et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 2016, 21, 1112–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans-Galea, M.V.; Carrodus, N.; Rowley, S.M.; Corben, L.A.; Tai, G.; Saffery, R.; Galati, J.C.; Wong, N.C.; Craig, J.M.; Lynch, D.R.; et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012, 71, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.I.; King, O.D.; Himeda, C.L.; Homma, S.; Chen, J.C.J.J.; Beermann, M.L.; Yan, C.; Emerson, C.P.; Miller, J.B.; Wagner, K.R.; et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin. Epigenetics 2015, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.L.F.; Goeman, J.J.; van der Vliet, P.J.; van Nieuwenhuizen, M.P.; Balog, J.; Vos-Versteeg, M.; Camano, P.; Ramos Arroyo, M.A.; Jerico, I.; Rogers, M.T.; et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015, 24, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, M.-C.; Roche, S.; Dion, C.; Tasmadjian, A.; Bouget, G.; Salort-Campana, E.; Vovan, C.; Chaix, C.; Broucqsault, N.; Morere, J.; et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014, 83, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Calandra, P.; Cascino, I.; Lemmers, R.J.L.F.L.F.; Galluzzi, G.; Teveroni, E.; Monforte, M.; Tasca, G.; Ricci, E.; Moretti, F.; Van Der Maarel, S.M.; et al. Allele-specific DNA hypomethylation characterises FSHD1 and FSHD2. J. Med. Genet. 2016, 53, 348–355. [Google Scholar] [CrossRef]
- Nikolic, A.; Jones, T.I.; Govi, M.; Mele, F.; Maranda, L.; Sera, F.; Ricci, G.; Ruggiero, L.; Vercelli, L.; Portaro, S.; et al. Interpretation of the epigenetic signature of facioscapulohumeral muscular dystrophy in light of genotype-phenotype studies. IJMS. under review.
- Roche, S.; Dion, C.; Broucqsault, N.; ere, C.; Gaillard, M.-C.; Robin, J.D.; Lagarde, A.; Puppo, F.; Vovan, C.; Chaix, C.; et al. Methylation hotspots evidenced by deep sequencing in patients with facioscapulohumeral dystrophy and mosaicism. Neurol. Genet. 2019, 5, e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.E.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, M.-C.; Puppo, F.; Roche, S.; Dion, C.; Campana, E.S.; Mariot, V.; Chaix, C.; Vovan, C.; Mazaleyrat, K.; Tasmadjian, A.; et al. Segregation between SMCHD1 mutation, D4Z4 hypomethylation and Facio-Scapulo-Humeral Dystrophy: A case report. BMC Med. Genet. 2016, 17, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T. Whole-genome methylation scan in ICF syndrome: Hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum. Mol. Genet. 2000, 9, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Dion, C.; Roche, S.; Laberthonnière, C.; Broucqsault, N.; Mariot, V.; Xue, S.; Gurzau, A.D.; Nowak, A.; Gordon, C.T.; Gaillard, M.-C.; et al. SMCHD1 is involved in de novo methylation of the DUX4 -encoding D4Z4 macrosatellite. Nucleic Acids Res. 2019, 47, 2822–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balog, J.; Goossens, R.; Lemmers, R.J.L.F.; Straasheijm, K.R.; Van Der Vliet, P.J.; Van Den Heuvel, A.; Cambieri, C.; Capet, N.; Feasson, L.; Manel, V.; et al. Monosomy 18p is a risk factor for facioscapulohumeral dystrophy. J. Med. Genet. 2018, 55, 469–478. [Google Scholar] [CrossRef]
- Nguyen, K.; Walrafen, P.; Bernard, R.; Attarian, S.; Chaix, C.; Vovan, C.; Renard, E.; Dufrane, N.; Pouget, J.; Vannier, A.; et al. Molecular combing reveals allelic combinations in facioscapulohumeral dystrophy. Ann. Neurol. 2011, 70, 627–633. [Google Scholar] [CrossRef]
- Nguyen, K.; Puppo, F.; Roche, S.; Gaillard, M.-C.; Chaix, C.; Lagarde, A.; Pierret, M.; Vovan, C.; Olschwang, S.; Salort-Campana, E.; et al. Molecular combing reveals complex 4q35 rearrangements in Facioscapulohumeral dystrophy. Hum. Mutat. 2017, 38, 1432–1441. [Google Scholar] [CrossRef]
- Nguyen, K.; Broucqsault, N.; Chaix, C.; Roche, S.; Robin, J.D.; Vovan, C.; Gerard, L.; Mégarbané, A.; Urtizberea, J.A.; Bellance, R.; et al. Deciphering the complexity of the 4q and 10q subtelomeres by molecular combing in healthy individuals and patients with facioscapulohumeral dystrophy. J. Med. Genet. 2019, 56, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Lengerke, C.; Daley, G.Q. Disease models from pluripotent stem cells: Turning back time in disease pathogenesis. Ann. N. Y. Acad. Sci. 2009, 1176, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A. Modeling for Lesch-Nyhan Disease by Gene Targeting in Human Embryonic Stem Cells. Stem Cells 2004, 22, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Lowry, W.E.; Richter, L.; Yachechko, R.; Pyle, A.D.; Tchieu, J.; Sridharan, R.; Clark, A.T.; Plath, K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 2883–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental Study of Fragile X Syndrome Using Human Embryonic Stem Cells Derived from Preimplantation Genetically Diagnosed Embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Wu, Z.; Liu, Z.; Hu, G.; Yu, J.; Chang, K.H.; Kim, K.P.; Le, T.; Faull, K.F.; Rao, N.; et al. Selective demethylation and altered gene expression are associated with ICF syndrome in human-induced pluripotent stem cells and mesenchymal stem cells. Hum. Mol. Genet. 2014, 23, 6448–6457. [Google Scholar] [CrossRef] [Green Version]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef] [Green Version]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef]
- Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934. [Google Scholar] [CrossRef]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; vanderMaarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 Activates Germline Genes, Retroelements, and Immune Mediators: Implications for Facioscapulohumeral Dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral Dystrophy: Incomplete Suppression of a Retrotransposed Gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechner, U.; Nolte, J.; Wolf, M.; Shirneshan, K.; Hajj, N.E.; Weise, D.; Kaltwasser, B.; Zovoilis, A.; Haaf, T.; Engel, W. Comparative methylation profiles and telomerase biology of mouse multipotent adult germline stem cells and embryonic stem cells. MHR Basic Sci. Reprod. Med. 2009, 15, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres Acquire Embryonic Stem Cell Characteristics in Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, R.M.; Blasco, M.A. Telomeres and telomerase in adult stem cells and pluripotent embryonic stem cells. Adv. Exp. Med. Biol. 2010, 695, 118–131. [Google Scholar] [PubMed]
- Zeng, W.; de Greef, J.C.; Chen, Y.-Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Chen, Y.-Y.; Newkirk, D.A.; Wu, B.; Balog, J.; Kong, X.; Ball, A.R.; Zanotti, S.; Tawil, R.; Hashimoto, N.; et al. Genetic and epigenetic characteristics of FSHD-associated 4q and 10q D4Z4 that are distinct from non-4q/10q D4Z4 homologs. Hum. Mutat. 2014, 35, 998–1010. [Google Scholar] [CrossRef]
- Jiang, G.; Yang, F.; van Overveld, P.G.M.; Vedanarayanan, V.; van der Maarel, S.; Ehrlich, M. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum. Mol. Genet. 2003, 12, 2909–2921. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, A.; Rival-Gervier, S.; Boussouar, A.; Foerster, A.M.; Rondier, D.; Sacconi, S.; Desnuelle, C.; Gilson, E.; Magdinier, F. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in Facio-Scapulo-Humeral dystrophy. PLoS Genet. 2009, 5, e1000394. [Google Scholar] [CrossRef]
- Bodega, B.; Ramirez, G.D.C.; Grasser, F.; Cheli, S.; Brunelli, S.; Mora, M.; Meneveri, R.; Marozzi, A.; Mueller, S.; Battaglioli, E.; et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Cortesi, A.; Pesant, M.; Sinha, S.; Marasca, F.; Sala, E.; Gregoretti, F.; Antonelli, L.; Oliva, G.; Chiereghin, C.; Soldà, G.; et al. 4q-D4Z4 chromatin architecture regulates the transcription of muscle atrophic genes in facioscapulohumeral muscular dystrophy. Genome Res. 2019, 29, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.; Pirozhkova, I.; Carnac, G.; Laoudj, D.; Lipinski, M.; Vassetzky, Y.S. Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystropny patients versus normal human myoblasts. Proc. Natl. Acad. Sci. USA 2006, 103, 6982–6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kollhoff, A.; Bergmann, A.; Stubbs, L. Methylation-sensitive binding of transcription factor YY1 to an insulator sequence within the paternally expressed imprinted gene, Peg3. Hum. Mol. Genet. 2003, 12, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Huichalaf, C.; Micheloni, S.; Ferri, G.; Caccia, R.; Gabellini, D. DNA methylation analysis of the macrosatellite repeat associated with FSHD muscular dystrophy at single nucleotide level. PLoS ONE 2014, 9, e115278. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef] [Green Version]
- Hartweck, L.M.; Anderson, L.J.; Lemmers, R.J.; Dandapat, A.; Toso, E.A.; Dalton, J.C.; Tawil, R.; Day, J.W.; Van Der Maarel, S.M.; Kyba, M. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology 2013, 80, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.; Allinne, J.; Pirozhkova, I.; Laoudj, D.; Lipinski, M.; Vassetzky, Y.S. A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: Implications for the facio-scapulo-humeral dystrophy. Genome Res. 2008, 18, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Gaszner, M.; Felsenfeld, G. Insulators: Exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006, 7, 703–713. [Google Scholar] [CrossRef]
- Ghirlando, R.; Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 2016, 30, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, A.; Ricci, G.; Sera, F.; Bucci, E.; Govi, M.; Mele, F.; Rossi, M.; Ruggiero, L.; Vercelli, L.; Ravaglia, S.; et al. Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1-3 D4Z4 reduced alleles: Experience of the FSHD Italian National Registry. BMJ Open 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shaw, N.D.; Brand, H.; Kupchinsky, Z.A.; Bengani, H.; Plummer, L.; Jones, T.I.; Erdin, S.; Williamson, K.A.; Rainger, J.; Stortchevoi, A.; et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 2017, 49, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.T.; Xue, S.; Yigit, G.; Filali, H.; Chen, K.; Rosin, N.; Yoshiura, K.I.; Oufadem, M.; Beck, T.J.; McGowan, R.; et al. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat. Genet. 2017, 49, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, G.; Cammish, P.; Siciliano, G.; Tupler, R.; Lochmuller, H.; Evangelista, T. Phenotype may predict the clinical course of facioscapolohumeral muscular dystrophy. Muscle Nerve 2019, 59, 711–713. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
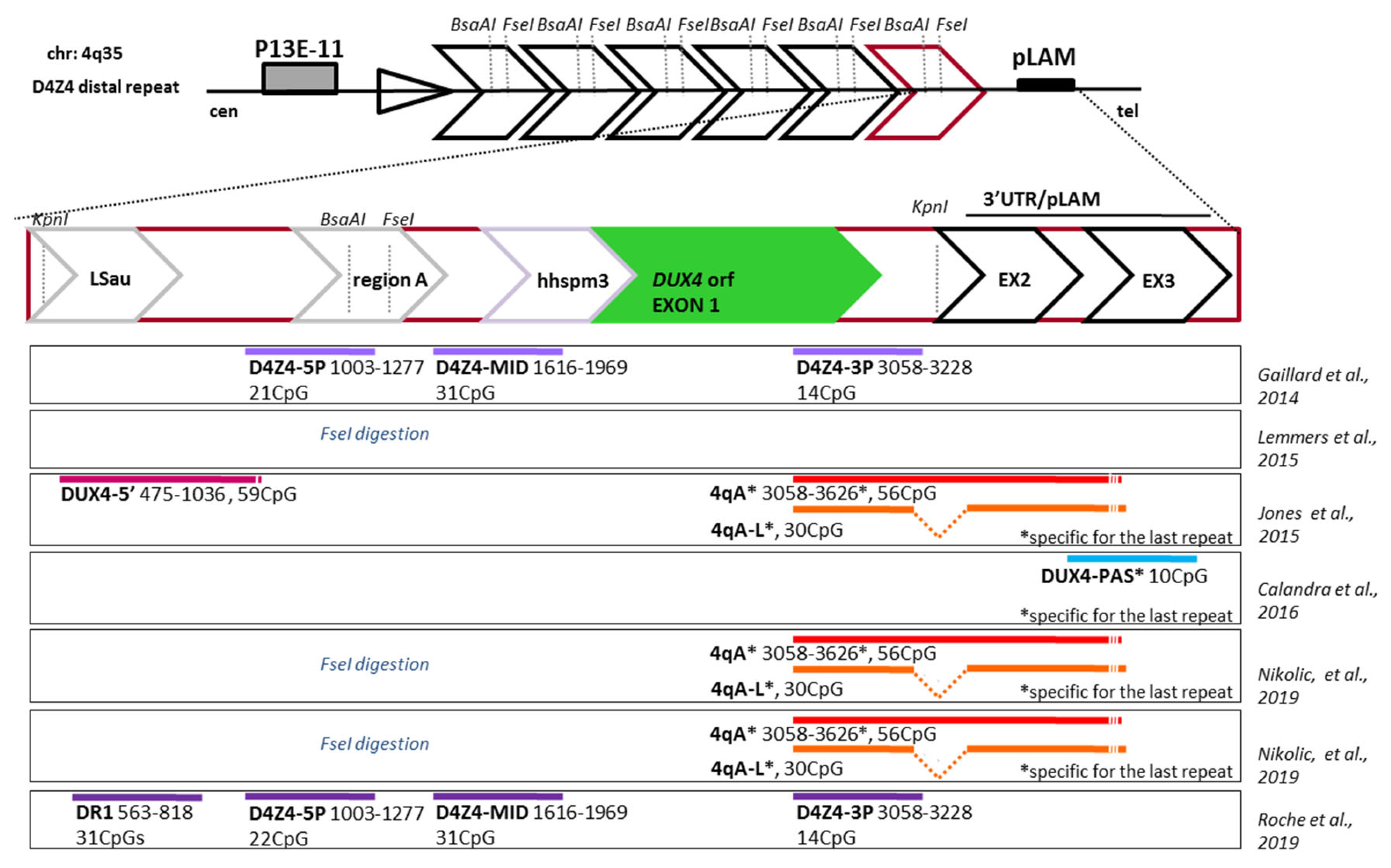
| REFs | Method | Assayed Region (s) | 4q- Specific | Nr.r of Subjects | Clinical Status (CCEF/ACSS) | Conclusions | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hypomethylation in FSHD1 /FSHD2 | Position within the Locus in which Hypomethylation was Found to be Discriminant | Correlation with D4Z4 Size | Correlation with Disease Severity | Matters | ||||||
| [69] | BSS | 4qA; 56 CpGs or 4qAL; 30 CpGs DUX4 5’; 59CpGs | Yes No | Famil:ies 12 Controls 10 Asymptomatic 22 FSHD1 | Not reoprted | Yes; Asymptomatyc carriers with intermediate methylation levels. | 4qA: pLAM only No DUX4 5’ | No | Not tested | .Different results using 5’ primers set. .Small differences between pathogenic contracted) and non-pathogenic allele. .No correlation with allele- specific % of DUX4 expression. |
| [70] | MRSE1 | FseI site | No | 254 controls ? 25 Asymptomatic 186 FSHD1 individuals 74 FSHD2-(just SMCHD1 mut) | ASCC | Yes; Asymptomatyc carriers not clearly defined | FseI site | Yes for 1–6 repeats carriers No for 7–10 repeats carriers | No for FSHD1; Yes for FSHD2 | .Unclear interpretation of data due to a global estimation of methylation as a function of D4Z4 repeat lengths. .Non-penetrant carriers of a DRA included in analysis. |
| [71] | meDIP BSS | D4Z4-5P; 22 CpGs D4Z4-MID; 31 CpGs D4Z4-3P; 14 CpGs | No | 20 Controls 29 Asymptomatic 37 FSHD1 9 FSHD2 8 FSHD1 4FSHD2 1Asymptomatic 7 Controls | Not reported | Yes; Asymptomatyc carriers with methylation levels not different than controls | D4Z4-5’ only No MID or 3’ | No | No | .Limited BSS analysis. No correlation with the global number of residual repeats. |
| [72] | BSS MRSE1 | DUX4-PAS FseI site | Yes No | 51 Controls 2Asymptomatic 44FSHD1 17 FSHD2 (just SMCHD1 mut) | ASCC | Yes; Asymptomatyc carriers not clearly defined No | DUX4-PAS: pLAM only No FseI | Yes | Yes | .Arbitrary selection of just one (number 6) CpG for statistical analysis. .Non-penetrant carriers of a DRA included in analysis. |
| [73] | MRSE1+ MRSE2 BSS+ MRSE1+ MRSE2 | FseI site BsaAI site 4qA; 56 CpGs or 4qAL; 30 CpGs | No Yes Yes | 122P+110: 88 FSHD1 P + 47FSHD1R 34 FSHD2P+ 2 FSHD 2 R 61 Asymptomatic R 1 Control 11FSHD1 8 Asymptomatic | CCEF | Assumed: beyond the purpose of the paper. Asymptomatyc carriers with methylation levels not different than index cases | FseI site 4qA: pLAM | No | No | -Limited BSS analysis |
| [74] | BSS +NGS | D4Z4-5P; 22 CpGs D4Z4-MID; 31 CpGs D4Z4-3P; 14CpGs DR1; 31CpGs 4aAint;14 CpGs 4qAext;42 CpGs | No Yes | 10 Controls 29 FSHD1 10 FSHD2-(just SMCHD1 mut) | Not reported | Yes Asymptomatyc carriers not included in analysis | D4Z4-5’ only | No | No | .Non-penetrant carriers of a DRA not included in analysis. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salsi, V.; Magdinier, F.; Tupler, R. Does DNA Methylation Matter in FSHD? Genes 2020, 11, 258. https://doi.org/10.3390/genes11030258
Salsi V, Magdinier F, Tupler R. Does DNA Methylation Matter in FSHD? Genes. 2020; 11(3):258. https://doi.org/10.3390/genes11030258
Chicago/Turabian StyleSalsi, Valentina, Frédérique Magdinier, and Rossella Tupler. 2020. "Does DNA Methylation Matter in FSHD?" Genes 11, no. 3: 258. https://doi.org/10.3390/genes11030258
APA StyleSalsi, V., Magdinier, F., & Tupler, R. (2020). Does DNA Methylation Matter in FSHD? Genes, 11(3), 258. https://doi.org/10.3390/genes11030258