Coding-Gene Coevolution Analysis of Rotavirus Proteins: A Bioinformatics and Statistical Approach




Abstract
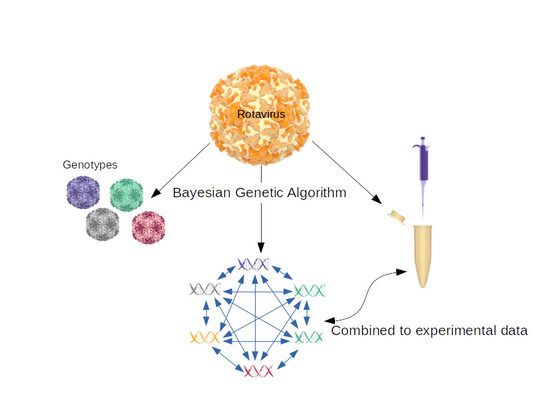
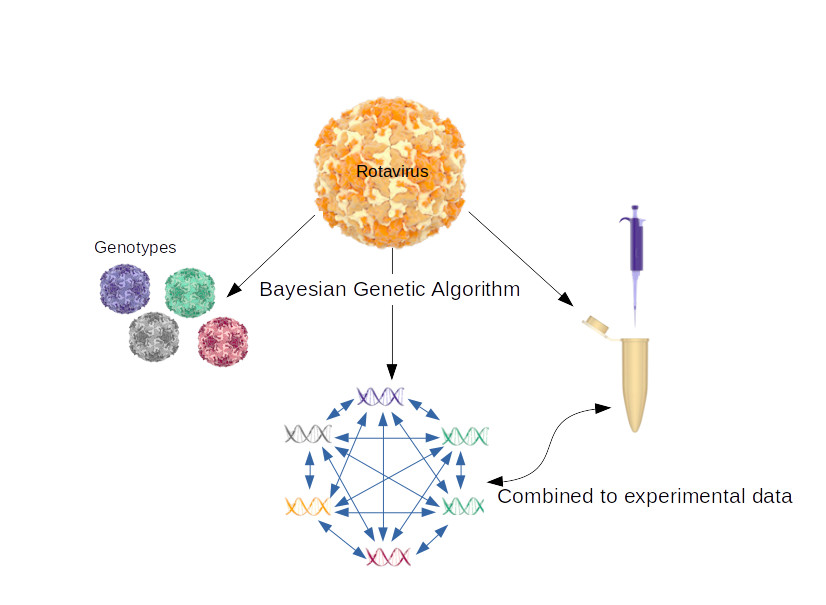
:
1. Introduction
2. Materials and Methods
2.1. Sequence Sampling
2.2. Sequence Alignment and Annotation
2.3. Coevolving Protein Residues and Calculation of Codon dN/dS Values
3. Results
3.1. Recombination and the Choice of the Model of Evolution
3.2. Intermolecular Interactions of RVA Proteins
3.3. Mapping of the Interacting Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tate, J.E.; Burton, A.H.; Boschi-Pinto, C.; Steele, A.D.; Duque, J.; Parashar, U.D. The WHO-coordinated Global Rotavirus Surveillance Network. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 136–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iman, K.K.; Asmaa, F.M.; Abdelrazek, Y.D.; Mohamed, F.H. Isolation and identification of Rotavirus infection in diarrheic calves at El Gharbia Governorate. Glob. VET 2017, 18, 178–182. [Google Scholar] [CrossRef]
- Godoy, H.P.; Hoppe, I.B. Spatial distribution of rotavirus in calves in the state of São Paulo, Brazil: 2006 to 2010. Rev. Agro Mbiente Line 2018, 12, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Desselberger, U. Rotaviruses. Virus Res. 2014, 190, 75–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Banyai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group a rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- O’Ryan, M. Rotarix (RIX4414): An oral human rotavirus vaccine. Expert Rev. Vaccines 2007, 6, 11–19. [Google Scholar] [CrossRef]
- Global Rotavirus Information and Surveillance Bulletin. Available online: https://who.int/immunization/diseases/rotavirus/rota_info_surv_bulletin/en/ (accessed on 17 June 2019).
- Heiman, E.M.; McDonald, S.M.; Barro, M.; Taraporewala, Z.F.; Bar-Magen, T.; Patton, J.T. Group a human rotavirus genomics: Evidence that gene constellations are influenced by viral protein interactions. J. Virol. 2008, 82, 11106–11116. [Google Scholar] [CrossRef] [Green Version]
- Jere, K.C.; Mlera, L.; O’Neill, H.G.; Potgieter, A.C.; Page, N.A.; Seheri, M.L.; van Dijk, A.A. Whole genome analyses of African G2, G8, G9, and G12 rotavirus strains using sequence-independent amplification and 454(R) pyrosequencing. J. Med. Virol. 2011, 83, 2018–2042. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gomara, M.; Maes, P.; Patton, J.T.; et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [Green Version]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary dynamics of human rotaviruses: Balancing reassortment with preferred genome constellations. PLoS Pathog. 2009, 5, e1000634. [Google Scholar] [CrossRef] [PubMed]
- Esona, M.D.; Banyai, K.; Foytich, K.; Freeman, M.; Mijatovic-Rustempasic, S.; Hull, J.; Kerin, T.; Steele, A.D.; Armah, G.E.; Geyer, A.; et al. Genomic characterization of human rotavirus G10 strains from the African Rotavirus Network: Relationship to animal rotaviruses. Infect. Genet. Evol. 2011, 11, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Kattoura, M.D.; Chen, X.; Patton, J.T. The rotavirus RNA-binding protein NS35 (NSP2) forms 10S multimers and interacts with the viral RNA polymerase. Virology 1994, 202, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Strimmer, K.; von Haeseler, A. Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.; Abdo, Z.; Joyce, P.; Sullivan, J. Performance-based selection of likelihood models for phylogeny estimation. Syst. Biol. 2003, 52, 674–683. [Google Scholar] [CrossRef]
- Abdo, Z.; Minin, V.N.; Joyce, P.; Sullivan, J. Accounting for uncertainty in the tree topology has little effect on the decision-theoretic approach to model selection in phylogeny estimation. Mol. Biol. Evol. 2005, 22, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP* Phylogenetic Analysis Using Parsimony (* and Other Methods), version 4.0a164 (X86); Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Adachi, J.; Hasegawa, M. MOLPHY—Programs for Molecular Phylogenetics Based on Maximum Likelihood, version 2.3; Institute of Statistical Mathematics: Tokyo, Japan, 1996. [Google Scholar]
- Sidorov, I.A.; Reshetov, D.A.; Gorbalenya, A.E. SNAD: Sequence Name Annotation-based Designer. BMC Bioinform. 2009, 10, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codoñern, F.M.; Fares, M.A. Why should we care about molecular coevolution? Evol. Bioinform. Online 2008, 4, 29–38. [Google Scholar]
- Donlin, M.J.; Szeto, B.; Gohara, D.W.; Aurora, R.; Tavis, J.E. Genome-wide networks of amino acid covariances are common among viruses. J. Virol. 2012, 86, 3050–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, A.F.; Lewis, F.I.; Frost, S.D.; Kosakovsky Pond, S.L. Spidermonkey: Rapid detection of co-evolving sites using Bayesian graphical models. Bioinformatics 2008, 24, 1949–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, N.; Koller, D. Being Bayesian about network structure. A Bayesian approach to structure discovery in Bayesian networks. Mach. Learn. 2003, 50, 95–125. [Google Scholar] [CrossRef]
- Korber, B. HIV Signature and Sequence Variation Analysis. In Computational Analysis of HIV Molecular Sequences; Rodrigo, A.G., Learn, G.H., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; Volume 1, pp. 55–72. [Google Scholar] [CrossRef]
- Korber, B.T.; Farber, R.M.; Wolpert, D.H.; Lapedes, A.S. Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein: An information theoretic analysis. Proc. Natl. Acad. Sci. USA 1993, 90, 7176–7180. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [CrossRef]
- Ota, T.; Nei, M. Variance and covariances of the numbers of synonymous and nonsynonymous substitutions per site. Mol. Biol. Evol. 1994, 11, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Grishin, N.V. AL2CO: Calculation of positional conservation in a protein sequence alignment. Bioinformatics 2001, 17, 700–712. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 1 April 2019).
- Jayaram, H.; Taraporewala, Z.; Patton, J.T.; Prasad, B.V. Rotavirus protein involved in genome replication and packaging exhibits a HIT-like fold. Nature 2002, 417, 311–315. [Google Scholar] [CrossRef]
- Taraporewala, Z.; Chen, D.; Patton, J.T. Multimers formed by the rotavirus nonstructural protein NSP2 bind to RNA and have nucleoside triphosphatase activity. J. Virol. 1999, 73, 9934–9943. [Google Scholar] [PubMed]
- Taraporewala, Z.F.; Jiang, X.; Vasquez-Del Carpio, R.; Jayaram, H.; Prasad, B.V.; Patton, J.T. Structure-function analysis of rotavirus NSP2 octamer by using a novel complementation system. J. Virol. 2006, 80, 7984–7994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpio, R.V.; Gonzalez-Nilo, F.D.; Jayaram, H.; Spencer, E.; Prasad, B.V.; Patton, J.T.; Taraporewala, Z.F. Role of the histidine triad-like motif in nucleotide hydrolysis by the rotavirus RNA-packaging protein NSP2. J. Biol. Chem. 2004, 279, 10624–10633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Farsetta, D.L.; Nibert, M.L.; Harrison, S.C. RNA synthesis in a cage—Structural studies of reovirus polymerase lambda3. Cell 2002, 111, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Piron, M.; Delaunay, T.; Grosclaude, J.; Poncet, D. Identification of the RNA-binding, dimerization, and eIF4GI-binding domains of rotavirus nonstructural protein NSP3. J. Virol. 1999, 73, 5411–5421. [Google Scholar]
- Rajasekaran, D.; Sastri, N.P.; Marathahalli, J.R.; Indi, S.S.; Pamidimukkala, K.; Suguna, K.; Rao, C.D. The flexible C terminus of the rotavirus non-structural protein NSP4 is an important determinant of its biological properties. J. Gen. Virol. 2008, 89, 1485–1496. [Google Scholar] [CrossRef]
- Bowman, G.D.; Nodelman, I.M.; Levy, O.; Lin, S.L.; Tian, P.; Zamb, T.J.; Udem, S.A.; Venkataraghavan, B.; Schutt, C.E. Crystal structure of the oligomerization domain of NSP4 from rotavirus reveals a core metal-binding site. J. Mol. Biol. 2000, 304, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Ouldali, M.; Menetrey, J.; Poncet, D. Structural organisation of the rotavirus nonstructural protein NSP5. J. Mol. Biol. 2011, 413, 209–221. [Google Scholar] [CrossRef]
- Martin, D.; Charpilienne, A.; Parent, A.; Boussac, A.; D’Autreaux, B.; Poupon, J.; Poncet, D. The rotavirus nonstructural protein NSP5 coordinates a [2Fe-2S] iron-sulfur cluster that modulates interaction to RNA. FASEB J. 2013, 27, 1074–1083. [Google Scholar] [CrossRef]
- McDonald, S.M.; Aguayo, D.; Gonzalez-Nilo, F.D.; Patton, J.T. Shared and group-specific features of the rotavirus RNA polymerase reveal potential determinants of gene reassortment restriction. J. Virol. 2009, 83, 6135–6148. [Google Scholar] [CrossRef] [Green Version]
- McDonald, S.M.; Tao, Y.J.; Patton, J.T. The ins and outs of four-tunneled Reoviridae RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2009, 19, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, B.; Settembre, E.; Temple, B.R.; Bellamy, A.R.; Harrison, S.C. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 A resolution. J. Mol. Biol. 2010, 397, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, S.M.; Patton, J.T. Rotavirus VP2 core shell regions critical for viral polymerase activation. J. Virol. 2011, 85, 3095–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, K.M.; Snyder, M.J.; Dennis, A.F.; Patton, J.T. Predicted structure and domain organization of rotavirus capping enzyme and innate immune antagonist VP3. J. Virol. 2014, 88, 9072–9085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.M.; Chichon, F.J.; Martin-Forero, E.; Gonzalez-Camacho, F.; Carrascosa, J.L.; Caston, J.R.; Luque, D. New insights into rotavirus entry machinery: Stabilization of rotavirus spike conformation is independent of trypsin cleavage. PLoS Pathog. 2014, 10, e1004157. [Google Scholar] [CrossRef] [Green Version]
- Dormitzer, P.R.; Greenberg, H.B.; Harrison, S.C. Proteolysis of monomeric recombinant rotavirus VP4 yields an oligomeric VP5* core. J. Virol. 2001, 75, 7339–7350. [Google Scholar] [CrossRef] [Green Version]
- Dormitzer, P.R.; Nason, E.B.; Prasad, B.V.; Harrison, S.C. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 2004, 430, 1053–1058. [Google Scholar] [CrossRef]
- Mathieu, M.; Petitpas, I.; Navaza, J.; Lepault, J.; Kohli, E.; Pothier, P.; Prasad, B.V.; Cohen, J.; Rey, F.A. Atomic structure of the major capsid protein of rotavirus: Implications for the architecture of the virion. EMBO J. 2001, 20, 1485–1497. [Google Scholar] [CrossRef] [Green Version]
- Leena, M. Molecular Epidemiology of Human Rotaviruses—A Study in Genetic Diversity; University of Helsinki: Helsinki, Finland, 2001. [Google Scholar]
- Chen, J.Z.; Settembre, E.C.; Aoki, S.T.; Zhang, X.; Bellamy, A.R.; Dormitzer, P.R.; Harrison, S.C.; Grigorieff, N. Molecular interactions in rotavirus assembly and uncoating seen by high-resolution cryo-EM. Proc. Natl. Acad. Sci. USA 2009, 106, 10644–10648. [Google Scholar] [CrossRef] [Green Version]
- Green, K.Y.; Hoshino, Y.; Ikegami, N. Sequence analysis of the gene encoding the serotype-specific glycoprotein (VP7) of two new human rotavirus serotypes. Virology 1989, 168, 429–433. [Google Scholar] [CrossRef]
- Dyall-Smith, M.L.; Lazdins, I.; Tregear, G.W.; Holmes, I.H. Location of the major antigenic sites involved in rotavirus serotype-specific neutralization. Proc. Natl. Acad. Sci. USA 1986, 83, 3465–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreaux, C.E.; Kelly, D.F.; McDonald, S.M. Electron microscopic analysis of rotavirus assembly-replication intermediates. Virology 2015, 477, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poncet, D.; Laurent, S.; Cohen, J. Four nucleotides are the minimal requirement for RNA recognition by rotavirus non-structural protein NSP3. EMBO J. 1994, 13, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Mansell, E.A.; Patton, J.T. Comparative analysis of the rotavirus NS53 gene: Conservation of basic and cysteine-rich regions in the protein and possible stem-loop structures in the RNA. Virology 1993, 196, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.A.; Torres-Vega, M.A.; Lopez, S.; Arias, C.F. In vivo interactions among rotavirus nonstructural proteins. Arch. Virol. 1998, 143, 981–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; McDonald, P.W.; Thompson, T.A.; Dennis, A.F.; Akopov, A.; Kirkness, E.F.; Patton, J.T.; McDonald, S.M. Analysis of human rotaviruses from a single location over an 18-year time span suggests that protein coadaption influences gene constellations. J. Virol. 2014, 88, 9842–9863. [Google Scholar] [CrossRef] [Green Version]
- Uzri, D.; Greenberg, H.B. Characterization of rotavirus RNAs that activate innate immune signaling through the RIG-I-like receptors. PLoS ONE 2013, 8, e69825. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Pruijssers, A.J.; Dermody, T.S.; Garcia-Sastre, A.; Greenberg, H.B. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 2011, 85, 3717–3732. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.M. The Rotavirus Interferon Antagonist NSP1: Many Targets, Many Questions. J. Virol. 2016, 90, 5212–5215. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.W.; Mitzel, D.N.; Weisend, C.M.; Flenniken, M.L.; Hardy, M.E. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1. J. Virol. 2002, 76, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Patton, J.T. Rotavirus RNA replication requires a single-stranded 3’ end for efficient minus-strand synthesis. J. Virol. 1998, 72, 7387–7396. [Google Scholar] [PubMed]
- Hua, J.; Patton, J.T. The carboxyl-half of the rotavirus nonstructural protein NS53 (NSP1) is not required for virus replication. Virology 1994, 198, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Berois, M.; Sapin, C.; Erk, I.; Poncet, D.; Cohen, J. Rotavirus nonstructural protein NSP5 interacts with major core protein VP2. J. Virol. 2003, 77, 1757–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contin, R.; Arnoldi, F.; Campagna, M.; Burrone, O.R. Rotavirus NSP5 orchestrates recruitment of viroplasmic proteins. J. Gen. Virol. 2010, 91, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Torres-Vega, M.A.; Gonzalez, R.A.; Duarte, M.; Poncet, D.; Lopez, S.; Arias, C.F. The C-terminal domain of rotavirus NSP5 is essential for its multimerization, hyperphosphorylation and interaction with NSP6. J. Gen. Virol. 2000, 81, 821–830. [Google Scholar] [CrossRef]
- Arnoldi, F.; Campagna, M.; Eichwald, C.; Desselberger, U.; Burrone, O.R. Interaction of rotavirus polymerase VP1 with nonstructural protein NSP5 is stronger than that with NSP2. J. Virol. 2007, 81, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Halder, U.C.; Chattopadhyay, S.; Chanda, S.; Nandi, S.; Bagchi, P.; Nayak, M.K.; Chakrabarti, O.; Kobayashi, N.; Chawla-Sarkar, M. Rotaviral enterotoxin nonstructural protein 4 targets mitochondria for activation of apoptosis during infection. J. Biol. Chem. 2012, 287, 35004–35020. [Google Scholar] [CrossRef] [Green Version]
- Holloway, G.; Johnson, R.I.; Kang, Y.; Dang, V.T.; Stojanovski, D.; Coulson, B.S. Rotavirus NSP6 localizes to mitochondria via a predicted N-terminal a-helix. J. Gen. Virol. 2015, 96, 3519–3524. [Google Scholar] [CrossRef]
- Au, K.S.; Chan, W.K.; Burns, J.W.; Estes, M.K. Receptor activity of rotavirus nonstructural glycoprotein NS28. J. Virol. 1989, 63, 4553–4562. [Google Scholar]
- Maass, D.R.; Atkinson, P.H. Rotavirus proteins VP7, NS28 and VP4 form oligomeric structures. J. Virol. 1990, 64, 2632–2641. [Google Scholar]
- Sapin, C.; Colard, O.; Delmas, O.; Tessier, C.; Breton, M.; Enouf, V.; Chwetzoff, S.; Ouanich, J.; Cohen, J.; Wolf, C.; et al. Rafts promote assembly and atypical targeting of a nonenveloped virus, rotavirus, in Caco-2 cells. J. Virol. 2002, 76, 4591–4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, O.; Gardet, A.; Chwetzoff, S.; Breton, M.; Cohen, J.; Colard, O.; Sapin, C.; Trugnan, G. Different ways to reach the top of a cell. Analysis of rotavirus assembly and targeting in human intestinal cells reveals an original raft-dependent, Golgi-independent apical targeting pathway. Virology 2004, 327, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.G.; Martinez, J.L.; Hernandez, D.T.; Hernandez, H.O.; Perez-Delgado, A.; Mendez, M.; Wood, C.D.; Rendon-Mancha, J.M.; Silva-Ayala, D.; Lopez, S.; et al. Nanoscale organization of rotavirus replication machineries. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ding, K.; Celma, C.C.; Zhang, X.; Chang, T.; Shen, W.; Atanasov, I.; Roy, P.; Zhou, Z.H. In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nat. Commun. 2019, 10, 2216. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
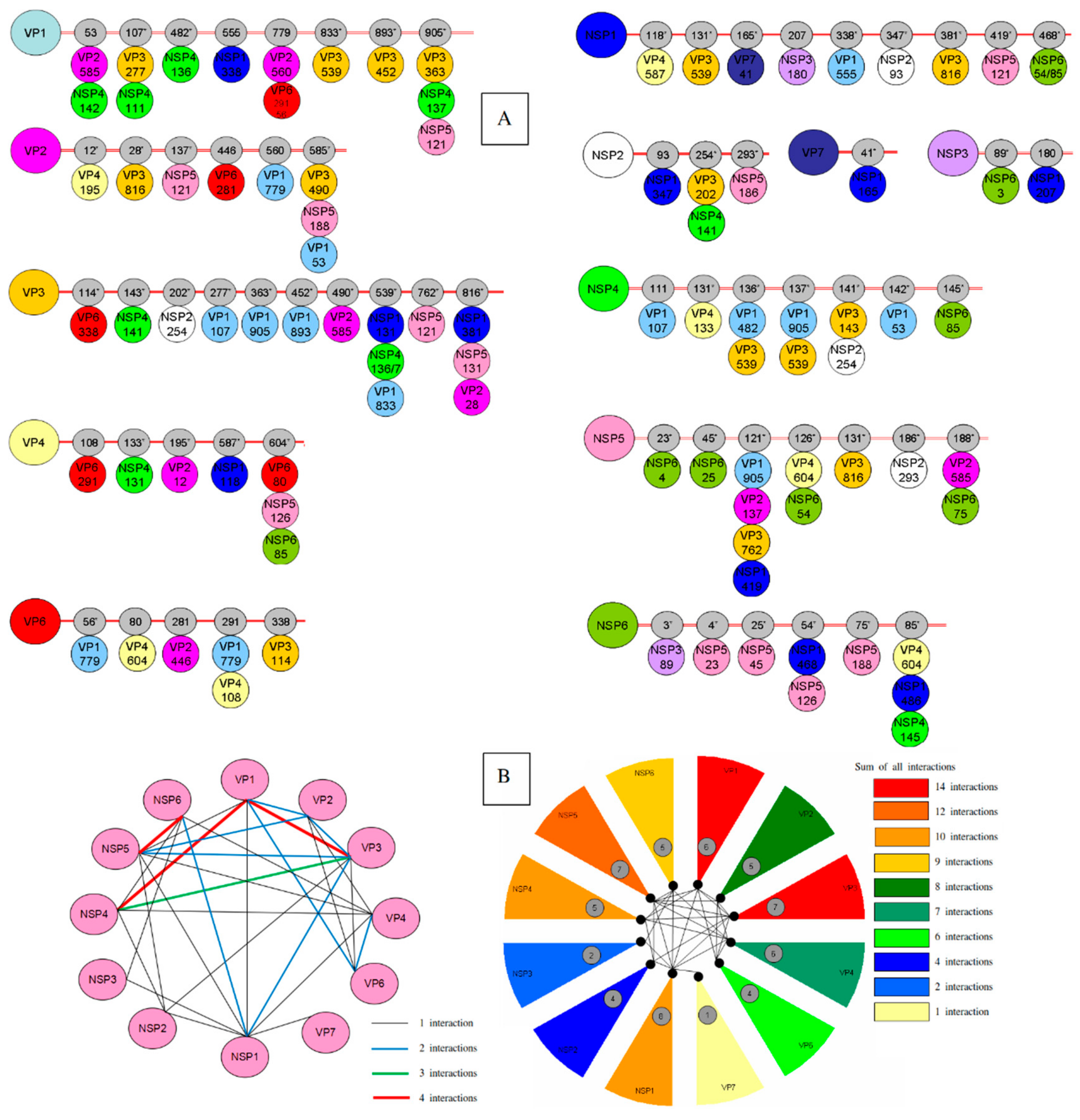
| Proteins | Residues (Intra-Coevolving Sites) | Residues (Inter-Coevolving Sites) |
|---|---|---|
| VP1 | 51*-53-107*-120-156*-293*-294*-357*-482*-555-657*-833*- 891*-1044* | 53-107*-482*-555-779-833*-893*-905* |
| VP2 | 12*-39*-40*-67*-128*-214*-229-446 | 12*-28*-137*-446-560-585* |
| VP3 | 54*-88-89-109*-114*-115*-116-143*-202*-203*-204*-205*-245*-266*-277*-336*-363*-373-405*-430*-437*-438-455*-478*-539*-625*-706*-707*-716-749*-751*-767*-798 | 114*-143*-202*-277*-363*-452*-490*-539*-762*-816* |
| VP4 | 106*-121*-131*-133*-135*-145*-150*-192-254*-280-283*-305*-337*-444*-586*-604*-630*-674*-713*-750* | 108-133*-195*-587*-604* |
| VP6 | 130*-199*-252 | 56*-80-281-291-338 |
| VP7 | 32-37*-50-57-68*-72*-73*-74*-94*-130-139*-149*-193-212*-237*-291*-303* | 41* |
| NSP1 | 10*-19*-55*-70*-93*-96*-108*-121*-163*-166*-180*-219*-223*-224*-225*-230*-253*-266*-268*-277*-293*-297*-312*-314*-326*-347*-357*-371*-372*-373*-381*-383*-388*-391*-402*-408*-419*-422*-435-436*-438*-440*-441*-459*-463*-476* | 118*-131*-165*-207-338*-347*-381*-419*-468* |
| NSP2 | 191-245*-256*-314* | 93-254*-293* |
| NSP3 | 186*-309* | 89*-180 |
| NSP4 | 141*-145*-148-169*-174 | 111-131*-136*-137*-141*-142*-145* |
| NSP5 | 112-177 | 23*-45*-121*-126*-131*-186*-188* |
| NSP6 | 75*-88* | 3*-4-25-54*-75*-85* |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abid, N.; Chillemi, G.; Salemi, M. Coding-Gene Coevolution Analysis of Rotavirus Proteins: A Bioinformatics and Statistical Approach. Genes 2020, 11, 28. https://doi.org/10.3390/genes11010028
Abid N, Chillemi G, Salemi M. Coding-Gene Coevolution Analysis of Rotavirus Proteins: A Bioinformatics and Statistical Approach. Genes. 2020; 11(1):28. https://doi.org/10.3390/genes11010028
Chicago/Turabian StyleAbid, Nabil, Giovanni Chillemi, and Marco Salemi. 2020. "Coding-Gene Coevolution Analysis of Rotavirus Proteins: A Bioinformatics and Statistical Approach" Genes 11, no. 1: 28. https://doi.org/10.3390/genes11010028
APA StyleAbid, N., Chillemi, G., & Salemi, M. (2020). Coding-Gene Coevolution Analysis of Rotavirus Proteins: A Bioinformatics and Statistical Approach. Genes, 11(1), 28. https://doi.org/10.3390/genes11010028