RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids, In Vitro Transcription, Cell Transfection, and Virus Quantification
2.3. Luciferase Assays
2.4. Virus Sequencing
2.5. One-Step Growth Analysis
2.6. Quantitative RT-PCR
2.7. Ribavirin-Sensitivity Assay
2.8. Cell-Based Recombination Assay
2.9. Polymerase Expression and Purification
2.10. Poly(rU) Polymerase Activity Assay
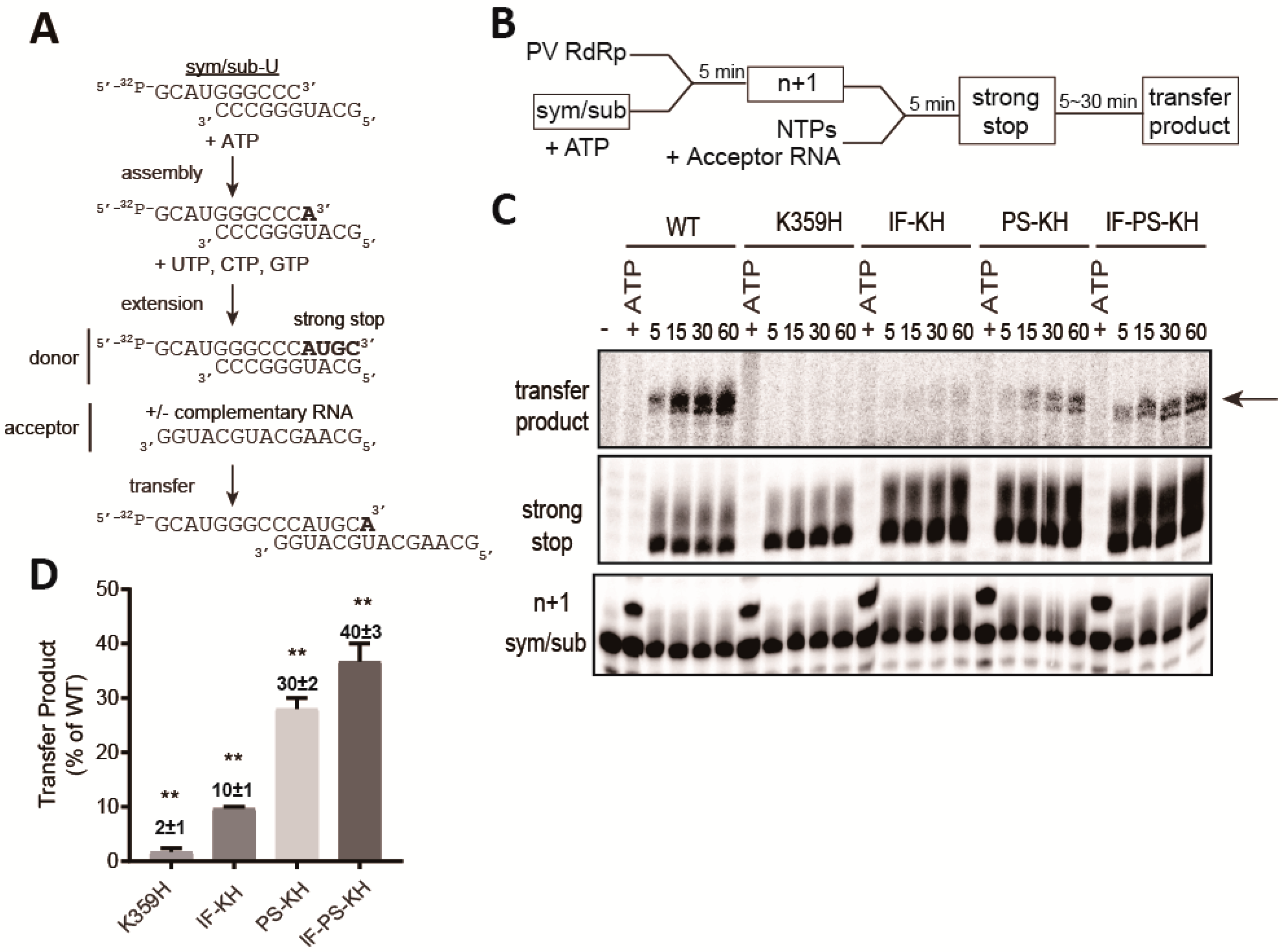
2.11. Sym/Sub-Based Template Switching Assay
3. Results
3.1. K359H PV Requires Two Second-Site Substitutions to Restore a “Wild-Type” Growth Phenotype
3.2. Characterization of the Double- and Triple-Mutant Viruses and Their Polymerases
3.3. Biochemical Properties of the RdRp Other Than Speed and Fidelity Contribute to the Efficiency of Recombination in Cell Culture
3.4. Poly(rU) Polymerase Activity as a Predictor of the Efficiency of Copy-Choice Recombination in Cell Culture
3.5. Polymerase Determinants Supporting Efficient Copy-Choice Recombination do not Overlap Completely With Determinants Supporting Efficient Forced-Copy-Choice Recombination
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- NIAID Emerging Infectious Diseases/Pathogens. Available online: https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens (accessed on 12 September 2019).
- Graci, J.D.; Cameron, C.E. Therapeutically targeting RNA viruses via lethal mutagenesis. Future Virol. 2008, 3, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Domingo, E. Antiviral Strategies Based on Lethal Mutagenesis and Error Threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Moustafa, I.M.; Arnold, J.J. Fidelity of Nucleotide Incorporation by the RNA-Dependent RNA Polymerase from Poliovirus. Enzymes 2016, 39, 293–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.A.; August, A.; Arnold, J.J.; Cameron, C.E. Polymerase Mechanism-Based Method of Viral Attenuation. Methods Mol. Biol. 2016, 1349, 83–104. [Google Scholar] [CrossRef]
- Weeks, S.A.; Lee, C.A.; Zhao, Y.; Smidansky, E.D.; August, A.; Arnold, J.J.; Cameron, C.E. A Polymerase mechanism-based strategy for viral attenuation and vaccine development. J. Biol. Chem. 2012, 287, 31618–31622. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef]
- Li, C.; Wang, H.; Yuan, T.; Woodman, A.; Yang, D.; Zhou, G.; Cameron, C.E.; Yu, L. Foot-and-mouth disease virus type O specific mutations determine RNA-dependent RNA polymerase fidelity and virus attenuation. Virology 2018, 518, 87–94. [Google Scholar] [CrossRef]
- Korboukh, V.K.; Lee, C.A.; Acevedo, A.; Vignuzzi, M.; Xiao, Y.; Arnold, J.J.; Hemperly, S.; Graci, J.D.; August, A.; Andino, R.; et al. RNA virus population diversity, an optimum for maximal fitness and virulence. J. Biol. Chem. 2014, 289, 29531–29544. [Google Scholar] [CrossRef]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.B.; Saleh, M.C.; Vignuzzi, M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wang, H.; Xie, X.; Li, C.; Zhou, G.; Yang, D.; Yu, L. Ribavirin-resistant variants of foot-and-mouth disease virus: The effect of restricted quasispecies diversity on viral virulence. J. Virol. 2014, 88, 4008–4020. [Google Scholar] [CrossRef] [PubMed]
- Gnadig, N.F.; Beaucourt, S.; Campagnola, G.; Borderia, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef]
- Coffey, L.L.; Beeharry, Y.; Borderia, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.J.; Vignuzzi, M.; Stone, J.K.; Andino, R.; Cameron, C.E. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J. Biol. Chem. 2005, 280, 25706–25716. [Google Scholar] [CrossRef]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, H.; Shi, J.; Yang, D.; Zhou, G.; Chang, J.; Cameron, C.E.; Woodman, A.; Yu, L. Senecavirus-Specific Recombination Assays Reveal the Intimate Link between Polymerase Fidelity and RNA Recombination. J. Virol. 2019. [Google Scholar] [CrossRef]
- Woodman, A.; Lee, K.M.; Janissen, R.; Gong, Y.N.; Dekker, N.H.; Shih, S.R.; Cameron, C.E. Predicting Intraserotypic Recombination in Enterovirus 71. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Long, J.C.; Bauer, D.L.V.; Fan, R.L.Y.; Yen, H.L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martin, M.J.; et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234–1242. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Nikolaitchik, O.A.; Keele, B.F.; Pathak, V.K.; Hu, W.S. Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity. Nucleic Acids Res. 2018, 46, 10535–10545. [Google Scholar] [CrossRef] [PubMed]
- Woodman, A.; Arnold, J.J.; Cameron, C.E.; Evans, D.J. Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 2016, 44, 6883–6895. [Google Scholar] [CrossRef] [PubMed]
- Poirier, E.Z.; Mounce, B.C.; Rozen-Gagnon, K.; Hooikaas, P.J.; Stapleford, K.A.; Moratorio, G.; Vignuzzi, M. Low-Fidelity Polymerases of Alphaviruses Recombine at Higher Rates to Overproduce Defective Interfering Particles. J. Virol. 2015, 90, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.; Smidansky, E.D.; Arnold, J.J.; Maksimchuk, K.R.; Moustafa, I.; Uchida, A.; Gotte, M.; Konigsberg, W.; Cameron, C.E. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat. Struct. Mol. Biol. 2009, 16, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Gohara, D.W.; Ha, C.S.; Kumar, S.; Ghosh, B.; Arnold, J.J.; Wisniewski, T.J.; Cameron, C.E. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr. Purif. 1999, 17, 128–138. [Google Scholar] [CrossRef]
- Gohara, D.W.; Crotty, S.; Arnold, J.J.; Yoder, J.D.; Andino, R.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol): Structural, biochemical, and biological analysis of conserved structural motifs A and B. J. Biol. Chem. 2000, 275, 25523–25532. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A bilirubin-inducible fluorescent protein from eel muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef]
- Oh, H.S.; Banerjee, S.; Aponte-Diaz, D.; Sharma, S.D.; Aligo, J.; Lodeiro, M.F.; Ning, G.; Sharma, R.; Arnold, J.J.; Cameron, C.E. Multiple poliovirus-induced organelles suggested by comparison of spatiotemporal dynamics of membranous structures and phosphoinositides. PLoS Pathog. 2018, 14, e1007036. [Google Scholar] [CrossRef]
- Kempf, B.J.; Peersen, O.B.; Barton, D.J. Poliovirus Polymerase Leu420 Facilitates RNA Recombination and Ribavirin Resistance. J. Virol. 2016, 90, 8410–8421. [Google Scholar] [CrossRef]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ‘imprecise’ intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J. Biol. Chem. 1999, 274, 2706–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severson, W.E.; Schmaljohn, C.S.; Javadian, A.; Jonsson, C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003, 77, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: Molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef]
- Sierra, M.; Airaksinen, A.; Gonzalez-Lopez, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: Implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Arias, A.; Arnold, J.J.; Sierra, M.; Smidansky, E.D.; Domingo, E.; Cameron, C.E. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J. Virol. 2008, 82, 12346–12355. [Google Scholar] [CrossRef] [Green Version]
- Pathak, H.B.; Ghosh, S.K.; Roberts, A.W.; Sharma, S.D.; Yoder, J.D.; Arnold, J.J.; Gohara, D.W.; Barton, D.J.; Paul, A.V.; Cameron, C.E. Structure-function relationships of the RNA-dependent RNA polymerase from poliovirus (3Dpol). A surface of the primary oligomerization domain functions in capsid precursor processing and VPg uridylylation. J. Biol. Chem. 2002, 277, 31551–31562. [Google Scholar] [CrossRef] [Green Version]
- Herold, J.; Andino, R. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 2000, 74, 6394–6400. [Google Scholar] [CrossRef] [Green Version]
- Andino, R.; Rieckhof, G.E.; Achacoso, P.L.; Baltimore, D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993, 12, 3587–3598. [Google Scholar] [CrossRef]
- Banerjee, S.; Aponte-Diaz, D.; Yeager, C.; Sharma, S.D.; Ning, G.; Oh, H.S.; Han, Q.; Umeda, M.; Hara, Y.; Wang, R.Y.L.; et al. Hijacking of multiple phospholipid biosynthetic pathways and induction of membrane biogenesis by a picornaviral 3CD protein. PLoS Pathog. 2018, 14, e1007086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulin, D.; Vilfan, I.D.; Berghuis, B.A.; Hage, S.; Bamford, D.H.; Poranen, M.M.; Depken, M.; Dekker, N.H. Elongation-Competent Pauses Govern the Fidelity of a Viral RNA-Dependent RNA Polymerase. Cell Rep. 2015, 10, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J. Biol. Chem. 2000, 275, 5329–5336. [Google Scholar] [CrossRef] [Green Version]
- Peliska, J.A.; Benkovic, S.J. Fidelity of in vitro DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Biochemistry 1994, 33, 3890–3895. [Google Scholar] [CrossRef]
- Peliska, J.A.; Benkovic, S.J. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science 1992, 258, 1112–1118. [Google Scholar] [CrossRef]
- Peliska, J.A.; Balasubramanian, S.; Giedroc, D.P.; Benkovic, S.J. Recombinant HIV-1 nucleocapsid protein accelerates HIV-1 reverse transcriptase catalyzed DNA strand transfer reactions and modulates RNase H activity. Biochemistry 1994, 33, 13817–13823. [Google Scholar] [CrossRef]
- Castro, C.; Smidansky, E.; Maksimchuk, K.R.; Arnold, J.J.; Korneeva, V.S.; Gotte, M.; Konigsberg, W.; Cameron, C.E. Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases. Proc. Natl. Acad. Sci. USA 2007, 104, 4267–4272. [Google Scholar] [CrossRef] [Green Version]
- Parsley, T.B.; Cornell, C.T.; Semler, B.L. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J. Biol. Chem. 1999, 274, 12867–12876. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.S.; Reddigari, S.R.; Nicklin, M.J.; Hammerle, T.; Wimmer, E. Purification and characterization of poliovirus polypeptide 3CD, a proteinase and a precursor for RNA polymerase. J. Virol. 1992, 66, 7481–7489. [Google Scholar]
- Muller, H.J. The Relation of Recombination to Mutational Advance. Mutat. Res. 1964, 106, 2–9. [Google Scholar] [CrossRef]
- Felsenstein, J. The evolutionary advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar] [PubMed]
- Chao, L. Fitness of RNA virus decreased by Muller’s ratchet. Nature 1990, 348, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Kempf, B.J.; Watkins, C.L.; Peersen, O.B.; Barton, D.J. Picornavirus RNA Recombination Counteracts Error Catastrophe. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Furio, V.; Moya, A.; Sanjuan, R. The cost of replication fidelity in an RNA virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10233–10237. [Google Scholar] [CrossRef] [Green Version]
- Elena, S.F.; Sanjuan, R. Adaptive value of high mutation rates of RNA viruses: Separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef] [Green Version]
- Furio, V.; Moya, A.; Sanjuan, R. The cost of replication fidelity in human immunodeficiency virus type 1. Proc. Biol. Sci. 2007, 274, 225–230. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence (5′-3′) |
|---|---|
| 3D EcoRI-R | AAT TAA TTC ATC GAT GAA TTC GGG CC |
| 3D BglII-F | GGC AAA GAA GTG GAG ATC TTG GAT GC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Ellis, V.D., III; Woodman, A.; Zhao, Y.; Arnold, J.J.; Cameron, C.E. RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination. Genes 2019, 10, 968. https://doi.org/10.3390/genes10120968
Kim H, Ellis VD III, Woodman A, Zhao Y, Arnold JJ, Cameron CE. RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination. Genes. 2019; 10(12):968. https://doi.org/10.3390/genes10120968
Chicago/Turabian StyleKim, Hyejeong, Victor D. Ellis, III, Andrew Woodman, Yan Zhao, Jamie J. Arnold, and Craig E. Cameron. 2019. "RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination" Genes 10, no. 12: 968. https://doi.org/10.3390/genes10120968
APA StyleKim, H., Ellis, V. D., III, Woodman, A., Zhao, Y., Arnold, J. J., & Cameron, C. E. (2019). RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination. Genes, 10(12), 968. https://doi.org/10.3390/genes10120968